Юношеская миоклоническая эпилепсия причины, симптомы, методы лечения и профилактики
Юношеская миоклоническая эпилепсия — разновидность болезни, которая диагностируется в возрасте от 7 до 21 года. Симптоматика подразумевает миоклонические приступы, затрагивающие парные системы, чаще это зрительный аппарат и плечи. Своевременное лечение обеспечивает больному ребёнку полноценную жизнь. Отличительная черта — симптоматика чаще возникает после пробуждения. Частота возникновения не связана с половой принадлежностью. Заболевание быстро поддаётся лечению.

Симптомы
Клинические признаки отличаются в зависимости от разновидности заболевания:
Статью проверил

Дата публикации: 24 Марта 2021 года
Дата проверки: 24 Марта 2021 года
Дата обновления: 15 Ноября 2021 года
Содержание статьи
Причины
Основная причина возникновения.ювениального миоклонического синдрома — это наследственный фактор. Также врачи выделяют другие причины прогрессирования болезни:
Также возможны ситуации, когда точную причину возникновения эпилептического синдрома у ребёнка не удаётся установить.
Стадии развития
Врачи выделяют несколько стадий прогрессирования болезни:
Диагностика
Назначаемые диагностические процедуры зависят от специфики миоклонической эпилепсии у больного. Чаще используют следующие методы:
При необходимости проводят суточное наблюдение за больным, врачи провоцируют возникновение приступа при помощи резкого пробуждения или яркого света.
Что такое юношеская миоклоническая эпилепсия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Агранович А. О., эпилептолога со стажем в 11 лет.

Определение болезни. Причины заболевания
Юношеской миоклонической эпилепсией (синдромом Янца) называют эпилептический синдром, который проявляется внезапными подёргиваниями в мышцах — миоклоническими приступами (от греч. «myos» — мышца, «klonos» — беспорядочное движение). Заболевание обычно развивается в подростковом возрасте.
Подёргивания в первую очередь возникают в мышцах верхнего плечевого пояса и рук. Сначала пациенты не обращают на них внимания, но со временем эпизоды возникают всё чаще и ухудшают качество жизни. Например, во время приступов из рук могут выпадать предметы. В дальнейшем появляются подёргивания ног, из-за которых человек может упасть.
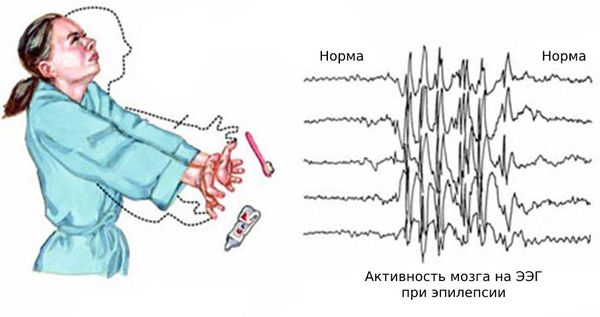
Нередко к этим эпизодам присоединяются генерализованные судорожные приступы — судороги возникают по всему телу и пациент теряет сознание. Также возникают абсансы — бессудорожные приступы с отключением сознания и амнезией на этот период. Как правило, частота генерализованных приступов невысокая: от одного за всю жизнь до раза в месяц. Подёргивания обычно случаются утром после пробуждения. Ярким провоцирующим фактором может стать недосыпание или вынужденное пробуждение. Также в трети случаев отмечается фотосенситивность — чувствительность к ритмическим вспышкам света.
Распространённость
Причины заболевания
Симптомы юношеской миоклонической эпилепсии
Чаще всего подёргивания возникают в верхнем плечевом поясе: мышцах рук и плеч с обоих сторон. Из-за этого пациенты нередко выпускают предметы из рук, например разбивают кружки и роняют зубные щётки. Однако возможны различные вариации миоклоний.
Приступы учащаются в утренние часы, особенно при недосыпе или вынужденном пробуждении.
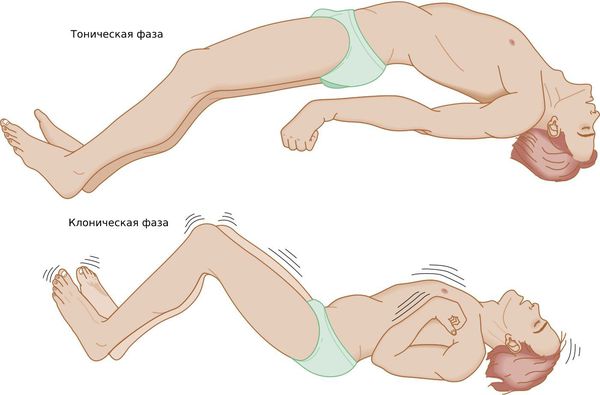
Генерализованный тонико-клонический приступ — состояние, при котором полностью отключается сознание. Приступ начинается с тонической фазы: напряжения в мышцах и специфического вскрикивания или хрипения. Руки полусогнуты и приподняты вверх или прижаты к телу. В этот момент из-за спазма дыхательной мускулатуры меняется цвет лица: оно синеет или сереет.
Далее развивается клоническая фаза, которая проявляется ритмичными подёргиваниями в конечностях. Она завершается полным мышечным расслаблением.
Патогенез юношеской миоклонической эпилепсии
Мозг человека состоит из двух основных типов клеток: нейронов и глии. Нейроны — это электрически возбудимые клетки, которые обрабатывают, хранят и передают информацию с помощью электрических и химических сигналов. Глиальные клетки играют в этом процессе вспомогательную роль.
Нейроны могут соединяться друг с другом и образовывать нервные сети. В пределах одного нейрона и его отростков информация передаётся в виде электрического возбуждения. В синапсе (месте контакта между нервными клетками) оно приводит к выделению различных химических веществ — нейромедиаторов.
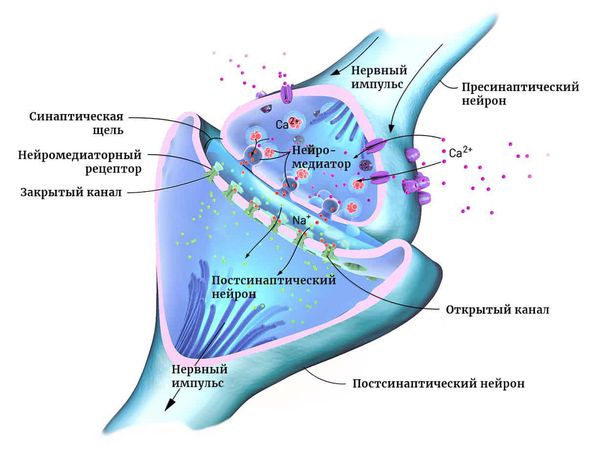
Нейромедиатор взаимодействует с рецепторами на мембране следующего нейрона. В результате в нём возникает электрическое возбуждение. Или не возникает — это зависит от конкретного нейромедиатора, активного в данный момент.
В нервных сетях между возбуждением и торможением работы нейронов поддерживается постоянный баланс. При сдвиге равновесия в сторону возбуждения происходит эпилептический приступ.
Классификация и стадии развития юношеской миоклонической эпилепсии
В 2017 году Международная лига борьбы с эпилепсией (ILAE) обновила классификацию заболевания, выделив четыре уровня диагностики:
1. Определить тип приступа: фокальный (возникающий из одного очага), генерализованный и с неизвестным началом. Миоклонические, тонико-клонические приступы и абсансы относятся к генерализованным приступам.
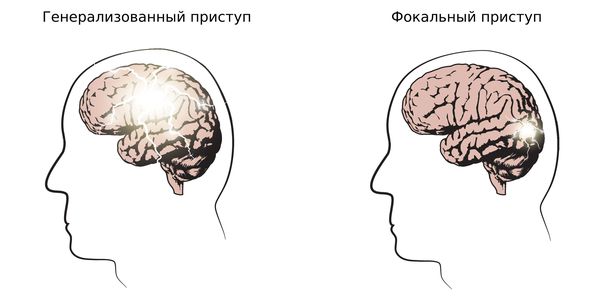
2. Установить тип эпилепсии: фокальная, генерализованная, сочетанная (фокальная + генерализованная) и неизвестная. Юношеская миоклоническая эпилепсия относится к генерализованной эпилепсии.
4. Выявить причины заболевания: генетические, структурные, метаболические, иммунные, инфекционные и с неизвестной этиологией. Юношеская миоклоническая эпилепсия в большинстве случаев вызвана генетическими факторами.
Классификация юношеской миоклонической эпилепсии проводится в зависимости от течения заболевания. Главный критерий — это наличие миоклонических приступов. Также выделяют варианты течения с добавлением генерализованных судорожных приступов и/или абсансов.
Осложнения юношеской миоклонической эпилепсии
Пациенты часто не обращают внимания на патологические сокращения мышц, поэтому к неврологу и эпилептологу больной зачастую обращается после появления генерализованных тонико-клонических приступов. В результате противоэпилептические препараты назначают с опозданием. На фоне этого приступы могут учащаться и угрожать здоровью и жизни пациента травмами и утоплениями.
Диагностика юношеской миоклонической эпилепсии
Основной диагностический критерий заболевания — это наличие миоклонических приступов.
Сбор анамнеза
На приёме врач спрашивает о необычных внезапных состояниях:
Пациенты могут не обращать внимания на такие симптомы и считать их своей особенностью. Абсансы и генерализованные тонико-клонические приступы с потерей сознания, особенно во сне, они могут и вовсе забывать. Поэтому при сборе анамнеза важно выяснить обстоятельства приступа не только у самих пациентов, но и у родственников и очевидцев.
Электроэнцефалограмма (ЭЭГ)
Основным способом диагностики эпилепсии является электроэнцефалограмма — метод исследования, при котором регистрируется суммарная электрическая активность клеток коры головного мозга.
Сейчас диагноз «эпилепсия» устанавливают с помощью длительного видео-ЭЭГ мониторинга — электроэнцефалограмма записывается параллельно с одной или несколькими видеокамерами, датчиком ЭКГ и при необходимости дополнительным контролем мышечной активности, частоты и глубины дыхания.
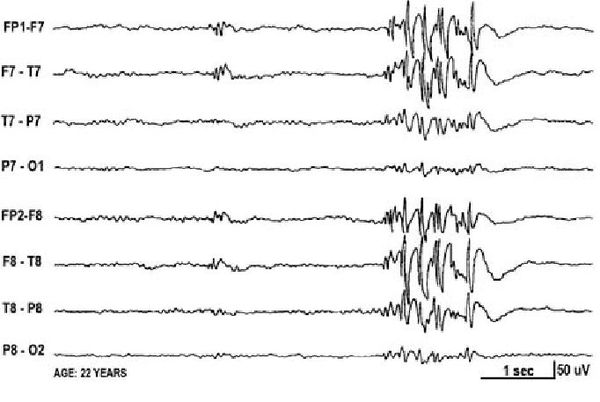
Основной фон биоэлектрической активности при юношеской миоклонической эпилепсии, как правило, соответствует возрастной норме. Патологическая активность проявляется короткими и генерализованными разрядами полиспайков (островолновых комплексов), которые регистрируются при миоклонических вздрагиваниях и полипик-волновыми комплексами между приступами.
Эпилептическая фотосенситивность — это предрасположенность к приступам под влиянием света. Может протекать бессимптомно или проявляться эпилептическими приступами под воздействием провоцирующих факторов: видеоигр, работы за компьютером, просмотра телевизора, мигающего освещения в ночных клубах и света природного происхождения.
Интеллект и неврологический статус при заболевании находятся в норме. Выражена эмоциональная неустойчивость и признаки невротического развития личности: резкая смена настроения, вспыльчивость и повышенная тревожность
Лечение юношеской миоклонической эпилепсии
Образ жизни
Антиэпилептические препараты

Ранее лидерами в лечении юношеской миоклонической эпилепсии являлись препараты вальпроевой кислоты. Они эффективны для прекращения приступов, но вызывают много побочных эффектов:
Помимо перечисленных препаратов, могут применяться «Топирамат», «Зонисамид», «Перампанел» и «Фенобарбитал».
Прогноз. Профилактика
Эффективность АЭП в предотвращении приступов достигает 90 %. При отмене терапии часто возникают рецидивы, поэтому потребуется длительный приём препаратов, иногда пожизненный.
Качество жизни значительно ухудшается при частых миоклонических и генерализованных тонико-клонических приступах, при которых пациенты рискуют получить травмы.
Профилактика
Особое внимание стоит уделить образу и режиму жизни пациента. Самыми мощными провоцирующими факторами являются недосыпание и злоупотребление алкоголем. А учитывая, что дебют заболевания приходится на подростковый возраст, молодые люди часто нарушают эти рекомендации, особенно в студенческие годы.
Пациент, у которого выявили фотосенситивность, предрасположен к приступам под воздействием мерцающего света. Поэтому им необходимо ограничить просмотр телевизора и работу за компьютером, исключить видеоигры и избегать посещения ночных клубов.
У всех пациентов с эпилепсией имеются определённые социальные ограничения: они не могут работать в некоторых сферах, водить автомобиль и нести военную службу. Все они определяются индивидуально соответствующими комиссиями.
МИОКЛОНУС-ЭПИЛЕПСИЯ
Миоклонус-эпилепсия (греч. mys, my [os] мышца + клонус; эпилепсия; син.: прогрессирующая миоклоническая эпилепсия, болезнь Унферрихта — Лундборга) — наследственное семейное заболевание центральной нервной системы, основным клиническим проявлением к-рого является сочетание миоклонического гиперкинеза с эпилептическими припадками. Описана Унферрихтом (H. Unverricht) в 1891 г. и Лундборгом (H. В. Lundborg) в 1903 г. как отдельная нозологическая форма.
Содержание
Этиология и патогенез
Заболевание наследуется по аутосомно-рецессивному типу. В основе его, по-видимому, лежит врожденное нарушение обмена веществ, но первичный биохимический дефект еще не выявлен. Оба пола заболевают одинаково часто. С. Н. Давиденков (1936) считал, что сложная картина Миоклонус-эпилепсии является выражением своеобразного плейотропизма гена этой болезни. Родители больных Миоклонус-эпилепсией могут быть здоровыми (нередки случаи их кровного родства). У родственников больных М.-э. наблюдаются и другие заболевания ц. н. с. — эпилепсия, паркинсонизм, хорея, умственная отсталость, алкоголизм и др. Описаны изоляты с относительно высокой частотой М.-э. Наряду с семейными наблюдается немало спорадических случаев.
Патологическая анатомия
При патологоанатомическом исследовании в головном мозге обнаруживаются дистрофические изменения, наиболее выраженные в зубчатом ядре мозжечка, оливах продолговатого мозга, черном веществе, полосатом теле, таламусе (зрительном бугре), в разных участках коры мозга. Характерным, но не обязательным гистопатологическим признаком М.-э. является наличие одного-двух шарообразных амилоидных включений преимущественно в нервных клетках, к-рые были открыты Лафорой (G. R. Lafora, 1911) и названы его именем. Тельца Лафоры обнаруживаются не только в ганглиозных клетках головного и спинного мозга, но также и в корешках спинномозговых нервов, в периферических нервах, скелетных мышцах, печени, селезенке. Во многих нервных клетках имеются также значительные скопления липофусцина.
Клиническая картина
В большинстве случаев болезнь начинается в возрасте 10—15 лет. Для нее характерно сочетание миоклонического гиперкинеза с эпилептическими припадками. У одних больных первыми симптомами являются миоклонии (см.), а общие эпилептические припадки (см. Эпилепсия), обычно ночные, присоединяются позже, нередко спустя ряд лет. У других больных вначале наблюдаются только эпилептические припадки, без миоклонического гиперкинеза, к-рый появляется позже. Кроме больших эпилептических припадков с потерей сознания, клонико-тоническими судорогами, недержанием мочи, прикусом языка, могут возникать и другие проявления эпилепсии — малые припадки, абсансы (разновидность малого эпилептического припадка), сумеречные состояния и др.
При М.-э. миоклонический гиперкинез характеризуется большим разнообразием и имеет свои особенности. Первоначально возникающие клонические подергивания являются чаще изолированными, могут наблюдаться в одной мышце; преимущественно сокращается четырехглавая мышца бедра, с незначительным двигательным эффектом. В дальнейшем миоклонии становятся все более распространенными, захватывают мышцы конечностей, туловища, головы, увеличивается их частота и двигательный эффект.
В большинстве случаев наблюдаются миоклонии быстрого темпа, молниеносные, беспорядочные, неритмичные, асинхронные, нерегулярные. Они значительно уменьшаются и прекращаются в спокойном состоянии, в лежачем положении, отсутствуют во время сна. Их усиливают произвольные движения и эмоции, особенно отрицательные. Наблюдаются так наз. сенсоклонические реакции — неожиданно возникший яркий свет, громкий звук вызывают кратковременный миоклонический приступ в виде внезапного усиления мышечных сокращений в конечностях, туловище, лице с резкими общими вздрагиваниями.
Миоклонический гиперкинез проявляется в различной степени в разные дни — он может значительно затихать или, наоборот, усиливаться, в связи с чем сами больные отмечают у себя чередование «хороших» и «плохих» дней. Иногда у нек-рых больных появляются психотонические приступы, описанные Лундборгом. Они выражаются в том, что под влиянием эмоциональных воздействий у больного возникает кратковременное тоническое напряжение всей мускулатуры без утраты сознания.
Описаны редко встречающиеся сочетания миоклонического гиперкинеза с хореоатетозом (см. Гиперкинезы). Имеется тесная связь между миоклоническими гиперкинезами и эпилептическими припадками. Изо дня в день усиливающиеся миоклонии постепенно становятся особенно резко выраженными, на высоте которых больной на короткое время теряет сознание, появляются клонико-тонические судороги с прикусом языка и недержанием мочи. После окончания эпилептического припадка сразу исчезают миоклонии, обычно на один-два дня, а затем они появляются вновь и постепенно усиливаются. Миоклонус-эпилепсия имеет прогрессирующее течение. Из-за миоклоний у больных затруднена и постепенно становится невозможной ходьба и выполнение других произвольных движений, они не могут себя обслуживать, самостоятельно принимать пищу. У отдельных больных наблюдаются легкие мозжечковые расстройства (атаксия, интенционное дрожание), мышечная гипотония. Чувствительность сохранена. Парезов, рефлекторных нарушений не отмечается. В поздних стадиях болезни развивается Экстрапирамидная обездвиженность с клин, картиной паркинсонизма (см.), при этом исчезает миоклонический гиперкинез.
За небольшими исключениями, больные деградируют психически.
На ранних этапах заболевания отмечаются астеноневротические расстройства, в дальнейшем больные становятся все более вялыми, апатичными, снижается интерес к окружающему, ухудшается память, ослабевает сообразительность, критика, временами вялость сменяется раздражительностью, бывают состояния спутанности с галлюцинаторными переживаниями, у многих развивается тяжелая деменция (см. Слабоумие). При электроэнцефалографическом исследовании (см. Электроэнцефалография) часто обнаруживаются пароксизмы электрической активности высокой амплитуды. Световая стимуляция, гипервентиляция усиливают пароксизмальную электрическую активность мозга. В сыворотке крови больных часто обнаруживается уменьшение содержания мукополисахаридов. По данным нек-рых авторов, для М.-э. характерна также повышенная концентрация в крови аргининсукцининовой к-ты.
Диагноз
Диагноз ставится на основании сочетания в клин, картине миоклонического гиперкинеза с эпилептическими припадками. Для диагностики важное значение имеет семейный характер заболевания. Дифференциальный диагноз проводится с весьма сходной мозжечковой миоклонической диссинергией Ханта — наследственным, аутосомно-рецессивным заболеванием, при к-ром также наблюдаются миоклонический гиперкинез и эпилептические припадки, но, в отличие от М.-э., мозжечковые расстройства гораздо более выражены. В отличие от М.-э., при кожевниковской эпилепсии (см.) клонические судороги наблюдаются в определенной части тела и обычно не носят генерализованного характера. Особенно трудно установить ранний диагноз в спорадических случаях, когда заболевание проявляется неполным клин, синдромом — или эпилептическими припадками, или миоклоническим гиперкинезом. В этом случае имеет значение семейный анамнез, а также уменьшение содержания мукополисахаридов в сыворотке крови.
Лечение и Прогноз
Лечение симптоматическое. Применяется противосудорожная терапия, назначают фенобарбитал, бензонал, седуксен, хлоралгидрат. Последние два средства могут значительно уменьшить на короткое время миоклонический гиперкинез. Применение хлоралгидрата может вызвать привыкание к нему и быстрое развитие кахексии, Показаны также повторные длительные курсы лечения глутаминовой к-той.
Прогноз неблагоприятный. Средняя продолжительность болезни ок. 20 лет, нек-рые больные доживают до старческого возраста. Наиболее часто причиной смерти является нарастающая кахексия, нередко пневмонии и другие интеркуррентные заболевания.
Супругам, в семье к-рых есть больной Миоклонус-эпилепсией, для решения вопроса о рождении ребенка рекомендуется обратиться в медико-генетическую консультацию (см.).
Библиография: Давиденков С. Н. Миоклонус-эпилепсия, в кн.: Неврол. и генетика, под ред. С. Н. Давиденкова, т. 2, с. 195, М., 1936; Дзержинский В. Myoclonia Unverricht’a, Журн, невропат, и психиат., кн. 5-6, с. 293, 1910; Маккьюсик В. А. Наследственные признаки человека, пер. с англ., М., 1976; Мельников С. А. Миоклонус-эпилепсия как синдром при некоторых заболеваниях головного мозга, Журн, невропат, и психиат., т. 57, в. 6, с. 740, 1957, библиогр.; de Ajuriaguerra J., Sigwald J. et Piot C. Myoclonie-epilepsie familiale de type Unverricht, Presse med., p. 1813, 1954; Bogaert L. Sur l’epilepsie-myoclonie progressive d’Unverricht — Lundborg, Mschr. Psychiat. Neurol., Bd 118, S. 170, 1949, Bibliogr.; Davison Ch. a. Keshner M. Myoclonus epilepsy, Arch. Neurol. Psychiat. (Chic.), v. 43, p. 524, 1940; Gambetti P. a. o. Myoclonic epilepsy with lafora bodies, Arch. Neurol. (Chic.), v. 25, p. 483, 1971; Hallidaу A. M. Les differents types des myoclonies, Rev. neurol., t. 119, p. 135, 1968; Handbook of clinical neurology, ed. by P. J. Yinken a. G. W. Bruyn, v. 15, p. 121, 1974, v. 27, p. 171, Amsterdam a. o., 1976; Lafora G. R. u. Glueck B. Beitrag zur Histopathologie der myoclonischen Epilepsie, Z. ges. Neurol. Psychiat., Bd 6, S. 1, 1911, Bibliogr.; LundborgH. Die progressive Myoclonus-Epilepsie, Up-sala, 1903; Unverricht H. Die Myoclonie, L]3z.— Wien, 1891.
Эпилепсия в детском возрасте
Эпилепсия — общее название группы хронических пароксизмальных болезней головного мозга, проявляющихся повторными судорожными или другими (бессудорожными) стереотипными припадками, сопровождающихся разнообразными (патологическими) изменениями личности и сн
Часть 3. Начало статьи читайте в № 6, 8, 2014 год
Существует немало форм эпилепсии, встречающихся исключительно в детском или подростковом возрасте. Именно зависимость от возраста многих разновидностей эпилепсии является главным отличительным признаком эпилептологии детского возраста [1–4].
Эпилепсии и судорожные синдромы периода новорожденности
Хотя продолжительность неонатального периода невелика, целый ряд эпилептических синдромов свойственен именно для новорожденных детей [3–5].
Доброкачественные семейные приступы (судороги) новорожденных
Доброкачественная неонатальная эпилепсия (с аутосомно-доминатным типом наследования) трех типов, проявляющаяся в первые 7 дней жизни (начиная с трех суток). В семейном анамнезе обязательно фигурируют указания на наличие в прошлом судорог у членов семьи пациента (в неонатальном периоде). Связь припадков с уточненными врожденными нарушениями метаболизма не установлена. Доброкачественные семейные неонатальные приступы манифестируют в виде фокальных и мультифокальных или генерализованных тонико-клонических (судорожных) припадков. Указанные припадки характеризуются малой продолжительность (1–2 мин) и значительной частотой (20–30 эпизодов за сутки). Впоследствии, по прошествии от 1 до 3 недель, приступы самопроизвольно спонтанно купируются.
Доброкачественные несемейные судороги (приступы) новорожденных («припадки пятого дня»)
Эта эпилепсия с дебютом в раннем неонатальном периоде имеет также другое название (доброкачественные идиопатические неонатальные судороги). Болезнь впервые описана в конце 1970-х гг. Судорожные приступы развиваются у доношенных новорожденных детей, не имевших до этого признаков патологии со стороны ЦНС. Дебют приступов происходит к концу 1-й недели жизни (в 80–90% случаев — между 4-м и 6-м днями), а их пик приходится на 5-й день жизни (отсюда и название). Описываемые приступы обычно имеют вид мультифокальных клонических судорог, которым нередко сопутствуют апноэ. В большинстве случаев доброкачественные идиопатические неонатальные судороги длятся не более 24 ч (они всегда прекращаются по прошествии 15 дней после дебюта). В 80% случаев за время судорожного периода у новорожденных отмечается развитие эпилептического статуса [3–5].
Ранняя инфантильная эпилептическая энцефалопатия с паттерном «угнетение/вспышка» на ЭЭГ (синдром Отахары)
Ранняя инфантильная эпилептическая энцефалопатия — редкая болезнь, относящаяся к злокачественным формам эпилепсии детского возраста. Дебютирует обычно в периоде новорожденности (или в возрасте 1–3 мес). Болезнь характеризуется тоническими приступами, частота которых значительно варьирует (10–300 эпизодов за сутки). У детей отмечается быстрое формирование неврологического дефицита и задержка психического развития. Специфический паттерн «вспышка/угнетение» при электроэнцефалографии (ЭЭГ) представлен у детей c синдромом Отахары как в состоянии сна, так и при бодрствовании. При магнитно-резонансной томографии (МРТ) головного мозга у пациентов отмечаются грубые аномалии развития ЦНС. Среди детей с ранней инфантильной эпилептической энцефалопатией с паттерном «вспышка/угнетение» на ЭЭГ летальность к возрасту 1 года достигает 40–50%. В 4–6-месячном возрасте синдром Отахары может трансформироваться в синдром Веста [3–6].
Ранняя миоклоническая (эпилептическая) энцефалопатия
Описана J. Aicardi и F. Goutières (1978); дебютирует преимущественно в периоде новорожденности (иногда до 3-месячного возраста). В генезе болезни предполагается роль генетических факторов и некоторых «врожденных ошибок метаболизма» (пропионовая ацидурия, метилмалоновая ацидемия, болезнь мочи с запахом кленового сиропа и др.). Клинически проявляется частыми миоклоническими припадками. Последние обычно не ассоциированы с ЭЭГ-изменениями во время приступа, но в ряде случаев одновременно с миоклониями регистрируются эпилептиформные разряды «угнетение/вспышка». Миоклонии чаще бывают фрагментарными (легкие подергивания дистальных отделов конечностей, век или углов рта); одновременно могут отмечаться фокальные (парциальные) приступы, массивные миоклонии и тонические спазмы (изолированные или серийные — возникают к 3–4 месяцам). Появление у ребенка тонических спазмов заставляет предположить наличие атипичного синдрома Веста, но вскоре основные проявления болезни возобновляются и сохраняются на протяжении длительного времени. Фокальные припадки (сложные парциальные — с заведением глаз или автономными симптомами: апноэ, гиперемия лица; клонические судороги в разных участках тела и др.) становятся основным типом приступов при ранней миоклонической эпилептической энцефалопатии. При интериктальном ЭЭГ-исследовании у детей регистрируется паттерн «угнетение/вспышка», состоящий из разрядов продолжительностью 1–5 сек, чередующийся с почти изоэлектрическими периодами (длительностью 3–10 сек). Описываемый ЭЭГ-паттерн становится более отчетливым во время сна (особенно в фазе глубокого сна). Изначальный паттерн «угнетение/вспышка» по достижении возраста 3–5 мес сменяется атипичной гипсаритмией или мультифокальными пароксизмами, но в большинстве случаев это лишь транзиторный феномен. Болезнь сопровождается высокой летальностью или прогрессивным распадом психомоторных функций (вплоть до вегетативного статуса), хотя по мере увеличения возраста частота и выраженность фокальных приступов и миоклоний постепенно уменьшаются [3–5, 7].
Витамин В6-зависимая эпилепсия
Cравнительно редкое наследственное заболевание, характеризующееся фармакорезистентными судорогами. Относится к группе метаболически обусловленных эпилепсий. Развивается у новорожденных, матери которых длительно получали пиридоксин во время беременности, а также при специфическом наследственном дефекте метаболизма (с повышенной потребностью в витамине В6). Известны случаи дебюта пиридоксинзависимых судорог у детей старше 1 мес и даже на втором году жизни. Между приступами судорог дети остаются беспокойными, реагируют мышечными подергиваниями на внешние раздражения. Болезнь не поддается обычному противосудорожному лечению, но назначение витамина В6 в высоких дозах (25 мг/кг/сут) быстро приводит к нормализации состояния [3–5].
Злокачественные мигрирующие парциальные судороги (приступы) младенческого возраста
Чрезвычайно редко встречающийся эпилептический синдром, описанный G. Coppola и соавт. (1995). К настоящему времени сообщается всего о примерно 50 случаях болезни, зарегистрированных в различных странах мира. Злокачественные мигрирующие парциальные судороги в 50% случаев наблюдаются в первые дни жизни; остальные 50% приходятся на возраст 1–3 мес. При дебюте приступы носят фокальный клонический характер, а по прошествии нескольких недель они становятся мультифокальными, причем исключительно частыми и фармакорезистентными к терапии антиэпилептическими препаратами. При ЭЭГ-исследовании у детей выявляется выраженная многоочаговая эпилептическая активность; метаболических нарушений не обнаруживается, а МРТ-признаки патологических изменений отсутствуют. Паталогоанатомическое исследование позволило выявить в гиппокампе признаки нейрональной потери [1, 3, 5, 8].
Эпилепсии у детей первого года жизни (1–12 мес)
По достижении 1-месячного возраста число разновидностей эпилептических синдромов, специфичных для первого года жизни ребенка, практически не уступает таковому, свойственному периоду новорожденности.
Инфантильные спазмы (синдром Веста)
Этот вариант катастрофической эпилепсии (генерализованной) бывает симптоматическим (подавляющее большинство случаев) или криптогенным (10–20%). Он манифестирует у детей на первом году жизни (чаще между 3-м и 8-м месяцами). В классическом варианте синдром Веста характеризуется в момент приступа комбинацией сгибательных и разгибательных движений, то есть выраженными миоклоническими (салаамовыми) спазмами, иногда серийными короткими сгибательными движениями головы («кивки»). Инфантильные спазмы могут развиться как вследствие наличия различной неврологической патологии, так и без каких-либо очевидных предшествующих нарушений функций ЦНС. При инфантильных спазмах психомоторное развитие замедляется, в дальнейшем высока вероятность выраженного отставания в развитии. В 80% случаев при синдроме Веста обнаруживаются микроцефалия, признаки детского церебрального паралича, атонически-атактические нарушения и др. Отличительным электрофизиологическим признаком синдрома Веста является гипсаритмия (по данным ЭЭГ), которая имеет вид диффузных высоковольтажных пиков и медленных волн, располагающихся на дезорганизованном (медленном) фоне. Прогноз синдрома Веста определяется эффективностью проводимой терапии, но в целом малоблагоприятен [3–8].
Тяжелая миоклонус-эпилепсия младенческого возраста (синдром Драве)
Болезнь, описанная C. Dravet (1978, 1992), дебютирует на первом году жизни (между 2-м и 9-м мес), что нередко происходит вслед за развитием фебрильного эпизода, вскоре после вакцинации или перенесения инфекции. Синдром Драве характеризуется появлением генерализованных или односторонних клонических судорог (обычно на фоне гипертермии или лихорадки), что происходит на фоне предшествующего нормального психомоторного развития ребенка на протяжении первого года жизни. Постепенно (по прошествии нескольких недель или месяцев) у ребенка развиваются афебрильные миоклонические и парциальные (фокальные) припадки. Прогрессивное нарастание частоты миоклоний (изолированных или серийных) предшествует появлению у пациентов генерализованных припадков. У детей выявляются умеренные мозжечковые и пирамидные знаки, связанные с дефицитарностью грубой моторики и атаксией походки. Нарушения психомоторного развития впоследствии отмечаются у детей примерно до 4-летнего возраста. Нередко при синдроме Драве у детей развивается эпилептический статус (судорожный или миоклонический). Данные ЭЭГ на протяжении первого года жизни обычно остаются в пределах нормы, хотя у отдельных пациентов встречаются спонтанные фотоиндуцированные пик-волновые разряды. Впоследствии иктальные ЭЭГ-исследования при синдроме Драве характеризуются наличием миоклонических или клонических припадков (генерализованная пик-волновая или полипик-волновая активность). Генерализованные разряды усиливаются в состоянии релаксации; одновременно отмечаются фокальные и мультифокальные пики и острые волны. Традиционные и новые антиэпилептические препараты обычно не предотвращают рецидива приступов при синдроме Драве. Прогноз по интеллектуальному развитию при синдроме Драве всегда неблагоприятен [3–5, 8].
Идиопатические доброкачественные парциальные эпилепсии младенчества
Обычно дебютируют у детей в возрасте 3–20 месяцев (чаще между 5-м и 8-м мес). Впервые описаны K. Watanabe и соавт. (1987), вследствие чего изначально получили обобщающее название «синдром Ватанабе». Характеризуются проявлениями в виде сложных парциальных (фокальных) приступов и благоприятным прогнозом (элиминация эпилептических припадков в течение 3 мес после дебюта). В среднем число приступов составляет около 7; у части пациентов отмечаются исключительно сложные парциальные припадки, у других — только вторично-генерализованные, а примерно в половине случаев встречается их сочетание. Во время приступа для пациентов характерны снижение реакции на предъявляемые стимулы, остановка двигательной активности, умеренные судорожные подергивания, латеральное заведение глаз и цианоз. Основными клиническими признаками этой группы эпилепсий являются высокая встречаемость кластерных приступов, короткая продолжительность припадков, а также изначально нормальные показатели интериктального ЭЭГ-исследования (впоследствии у части детей могут обнаруживаться пароксизмальные разряды) [2, 3, 5, 6, 8].
Сходные с идиопатическими доброкачественными парциальными эпилепсиями младенчества, но исключительно семейные пароксизмальные состояния с дебютом на первом году жизни носят название «доброкачественные инфантильные семейные судороги». В 1997 г. были описаны сходные с ними случаи семейных эпилепсий с последующим формированием хореоатетоза — семейные судороги с хореатетозом [3–5, 8, 9].
Эпилепсии у детей раннего возраста (1–3 года)
Для детей раннего возраста (от 12 до 36 месяцев), в первую очередь, характерны cиндром Доозе, синдром Леннокса–Гасто, доброкачественная миоклонуc-эпилепсия младенческого возраста, синдром гемиконвульсий-гемиплегии, идиопатическая парциальная эпилепсия младенчества, абсансная эпилепсия раннего детства, электрический эпилептический статус медленно-волнового сна, ранний и поздний детский нейрональный липофусциноз (типы I и II).
Миоклоническая астатическая эпилепсия раннего детского возраста (cиндром Доозе)
Представляет собой эпилепсию c миоклонически-астатическими приступами (различной продолжительности). Приступы дебютируют в возрасте 1–5 лет. Чаще болезнь поражает мальчиков. Астатические и миоклонические приступы могут сочетаться, причем миоклонии возникают как до, во время, так и после астатического припадка. Приступы наступают внезапно и практически всегда сопровождаются падениями. Миоклонии отмечаются в виде различной выраженности симметричных подергиваний в руках и мышцах плеч пояса, что сочетается с наклоном головы («кивки»). Признаки утраты сознания у детей в момент приступа отсутствуют. До начала заболевания психомоторное развитие детей обычно соответствует норме. У части детей болезнь осложняется риском развития деменции (предположительно за счет развития эпилептического статуса абсансов). При ЭЭГ регистрируются генерализованные билатерально-синхронные комплексы пик-волн (3 и более за 1 сек, 2–4 Гц). Прогноз при миоклонически-астатической эпилепсии раннего детского возраста малоблагоприятен [3–6, 8].
Синдром Леннокса–Гасто, или миокинетическая эпилепсия раннего детства с медленными пик-волнами
Группа гетерогенной патологии с эпилептическими приступами (атоническими, тоническими, атипичными абсансами), интеллектуальной дефицитарностью и характерным ЭЭГ-паттерном. Как и при синдроме Веста, при синдроме Леннокса–Гасто выделяют симптоматический и криптогенный варианты болезни. Ранние формы дебютируют примерно с 2-летнего возраста. До 30% случаев представляют собой результат трансформации из синдрома Веста. Клинически синдром Леннокса–Гасто характеризуется миоклонически-астатическими припадками, салаамовыми судорогами (молниеносными кивательными), атипичными абсансами, тоническими приступами (чаще во сне). Могут встречаться генерализованные тонико-клонические, миоклонические и фокальные (парциальные) приступы. Для детей типична серийность припадков с изменениями сознания (ступор) и постепенным переходом в эпилептический статус. Помимо эпилептических приступов, в неврологическом статусе могут отмечаться церебральные парезы/параличи, а также атонически-астатические нарушения (до 40% пациентов). У детей происходит снижение интеллекта (различной степени), наблюдаются выраженные нарушения когнитивных функций. По ЭЭГ-данным типичны изменения фоновой активности в виде медленных пик-волн
В. М. Студеникин, доктор медицинских наук, профессор, академик РАЕ
ФГБУ «НЦЗД» РАМН, Москва