Прогрессирующая мышечная дистрофия Дюшенна

Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Общие сведения
Причины мышечной дистрофии Дюшенна
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы мышечной дистрофии Дюшенна
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
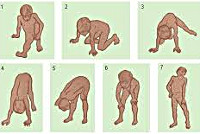
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика мышечной дистрофии Дюшенна
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК. При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
Лечение мышечной дистрофии Дюшенна
Терапия, применяемая в клинической практике, пока включает лишь необходимые симптоматические мероприятия. Для улучшения метаболизма мышечной ткани возможно назначение анаболических стероидов (метандиенона, нандролона деканоата), АТФ, актопротекторов (этилтиобензимидазола); для облегчения нервно-мышечной передачи — неостигмина. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия.
Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Поиск эффективных способов лечения дистрофии Дюшенна — задача, над решением которой трудятся сегодня специалисты в области неврологии, биохимии, генной инженерии. Из перспективных разработок в этом направлении можно выделить лечение стволовыми клетками, активацию гена утрофина, являющегося наиболее близким аналогом дистрофина, технологию пропуска экзонов.
Прогноз и профилактика
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Лечится ли миодистрофия Дюшенна?
Время чтения: 5 мин.
Лечится ли миодистрофия Дюшенна?
Миодистрофия Дюшенна встречается так же часто, как СМА (спинальная мышечная атрофия), но болеют практически только мальчики. Согласно статистике, один из 3500-5000 мальчиков в мире рождается с мышечной дистрофией Дюшенна.
Терапия СМА и Дюшенна: в чем разница?
При всей катастрофичности СМА молекулярная и клеточная основа этого заболевания проще и понятнее, чем у миодистрофии Дюшенна.
Лекарство можно ввести в спинной мозг, оно не размывается по всему организму, не метаболизируется печенью и не выводится почками. Создается его локальная высокая концентрация. Одной инъекции хватает на несколько месяцев. А если это генотерапия, то полноценный замещающий генетический материал, однажды попав в больную нервную клетку, вылечивает ее, остается там надолго, если не на всю жизнь.
В случае миодистрофии Дюшенна все сложнее: ген дистрофина — один из самых больших. Он больше гена СМА почти в полторы тысячи раз, и в нем тысячи разных мутаций (делеции, дупликации, нонсенс и т.д.) в разных местах.
Ген не помещается целиком в вирус, поэтому используют только кусочки гена — мини- и микродистрофин, которые могут ограниченно восстановить функциональность мышечных клеток, перевести «Дюшенн в Беккер» — более легкую форму миодистрофии, при которой человек может прожить до 60 лет, сохранять дееспособность, работать.
Мышечные клетки составляют 40% от всех клеток тела, они активно работают и постоянно заменяются. Доставить лекарство в мышечные клетки – трудная задача: оно должно с кровью попасть во все клетки, а раз с кровью, то с лекарством борется печень, и оно выводится через почки. Итог – низкая концентрация и ограниченное время действия.
Генотерапия мини- и микродистрофином – подводные камни на каждом этапе. У ребенка может быть имеющийся иммунитет к вирусу-переносчику гена, таких детей примерно 30%. Компании сейчас работают над тем, как убрать антитела к вирусу из крови. И пока пациентам доступна только одна попытка генотерапии в жизни, потому что после инфузии уже точно будет выраженный противовирусный иммунитет.
Но результат стоит того, тем более с осложнениями врачи научились бороться. И, конечно, будут другие технологии, лучше и безопаснее, которые смогут преодолеть существующие проблемы.
Больные МДД как снежинки: все разные
Сделать лекарство для МДД сложно, один препарат не вылечит всех, как при СМА.
Больные МДД как снежинки: все разные, нет одинаковых, уже описано около десяти тысяч мутаций гена. Имеет значение и вся генетика ребенка. Поэтому болезнь проявляется по-разному, даже в одной семье у двух братьев с одной мутацией.
| Препарат | Одобрено международными регуляторами | Ожидаемая дата регистрации в РФ | Возраст начала терапии |
|---|---|---|---|
| Аталурен | EMA 2012 | 24.11.2020 | 2 года |
| Экзондис 51 | FDA 2016 | 2021-22 | С момента постановки диагноза |
| Вайондис, Вилтепсо 53 | FDA 2020, Япония | 2021-22 | 4(NS) 0 (S) |
| Амондис | FDA 2021 | 2022 | С момента постановки диагноза |
| Мини/микродистрофин AAV вектор (4) | 2021-22 | 2 года | |
| Гивиностат | 2021-22 | ||
| Ваморолон | 2021-22 | 2 года | |
| Тамоксифен | Off-label | 2022 | |
| ГКСФ (гранулоцитарный колониестимулирующий фактор) | Off-label | 2022 |
Мы живем во время орфанной революции, когда многие редкие генетические неизлечимые заболевания получают патогенетической лечение, и больные обретают возможность жить долгой полноценной жизнью. Новые лекарства могут превратить фатальную болезнь в хроническую, хоть и тяжелую, как, например, диабет.
Как получить лекарства?
Если у вашего ребенка выявили миодистрофию Дюшенна, и вы хотите получить терапию для него, нужно прежде всего иметь подтвержденный генетический диагноз с определением мутации, которая вызвала заболевание. Примерно в 1% случаев не удается выяснить тип мутации. В таком случае делают биопсию мышцы и определяют количество синтезируемого мышцами дистрофина.
Как участвовать в клинических исследованиях?
Сейчас активно проводятся клинические исследования нескольких лекарственных препаратов. О том, какие из них исследуются в России, можно узнать на сайте Минздрава РФ. Заполнив форму, введя название препарата (Аталурен), можно узнать, в каком учреждении проводится исследование. Родителям необходимо самостоятельно связаться с учреждением, чтобы попробовать попасть в число участников.
Информацию о новых препаратах и клинических исследований в области миодистрофии Дюшенна можно найти и в пациентских сообществах, например, ProДюшенн.
Материал подготовлен с использованиюем гранта Президента Российской Федерации, предоставленного Фондом президентских грантов.
Использовано стоковое изображение от Depositphotos.
Прогрессирующая мышечная дистрофия Дюшшена/Беккера
Общая информация
Краткое описание
Прогрессирующая мышечная дистрофия Дюшенна/Беккера (OMIM – 310200) – наследственное рецессивное Х-сцепленное заболевание, с распространенностью 1 случай на 3600-6000 мальчиков, рожденных живыми, возникающее в результате мутации в гене дистрофина, характеризующееся поражением проксимальных групп мышц, кардиологическими, ортопедическими и респираторными осложнениями [1,2,3,4].
Соотношение кодов МКБ-10 и МКБ-9
| МКБ-10 | МКБ-9 | ||
| Код | Название | Код | Название |
| G 71.0 | Мышечная дистрофия · аутосомно-рецессивная детская, напоминающая дистрофию Дюшенна или Беккера; · доброкачественная [Беккера]; · злокачественная [Дюшенна]. | ||
Дата разработки/пересмотра протокола: 2016 год
Пользователи протокола: неврологи, педиатры, врачи общей практики, хирурги-ортопеды, кардиологи, клинические генетики.
Категория пациентов: дети.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортное или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
Молекулярно-генетическая:
Классификация нервно-мышечных заболеваний Вашингтонского университета [http://neuromuscular.wustl.edu]), основанная на первичном молекулярном дефекте.
Классификация прогрессирующих мышечных дистрофий (ПМД)
| Форма | Ген | Локализация |
| Х-сцепленные | ||
| ПМД Дюшенна | Дистрофин | Хр21 |
| ПМД Беккера | Дистрофин | Хр21 |
Диагностика (амбулатория)
ДИАГНОСТИКА НА АМБУЛАТОРНОМ УРОВНЕ
Диагностические критерии
Жалобы:
· мышечная слабость;
· повышенная утомляемость;
· частые падения;
· нарушение походки;
· затруднения при подъеме по лестнице, беге, ходьбе;
· ходьба на носочках;
· задержка речевого развития;
· нарушение поведения.
Анамнез:
· пол ребенка – мужской;
· дебют клинических проявлений в возрасте 3-5 лет (при ПМД Дюшенна), 8-10 лет (при ПМД Беккера);
· установленный диагноз «Прогрессирующая миодистрофия» у родственников мужского пола по материнской линии или неуточненное нервно-мышечное заболевание, наличие в семье женщин – носительниц патологического гена;
· задержка становления двигательных навыков и речи.
Физикальное обследование:
· оценка состояния нервно-мышечной системы с помощью стандартизированных шкал и тестов (таб.2);
· исследование мышечного тонуса и силы: проксимальный тип мышечных атрофий и парезов, слабость мышц тазового и плечевого пояса;
· исследование сухожильных рефлексов: снижение или утрата сухожильных рефлексов;
· исследование походки: неуклюжая, «утиная» походка;
· визуальный осмотр мышц: псевдогипертрофия икроножных и дельтовидных мышц;
· симптом Говерса;
· осмотр костно-суставной системы: контрактуры крупных суставов, поясничный лордоз, сколиоз, деформация грудной клетки и стоп;
· нейроповеденческие расстройства (СДВГ, аутистические проявления, обсессивно-компульсивные расстройства);
· непрогрессирующие отклонения в когнитивных функциях (нарушение кратковременной словесной памяти, дислексия, специфические расстройства обучения);
· эмоциональные расстройства (депрессия, тревожность).
Лабораторные исследования:
Биохимический анализ крови:
· повышение уровня КФК – является облигатным, ранним доклиническим признаком;
· повышение уровня трансаминаз: АЛТ, АСТ;
· повышение уровня ЛДГ- не является облигатным признаком.
Инструментальные исследования:
· УЗИ мышц – признаки мышечной дегенерации: замена мышечной ткани жировой или фиброзной тканью;
· ЭМГ – первично-мышечные изменения, миопатический тип ЭМГ: короткие остроконечные многочисленные потенциалы;
· ЭхоКГ – могут быть выявлены признаки гипертрофической или дилятационной кардиомиопатии.
Методы оценки состояния нервно-мышечной системы при ПМД Дюшенна/Беккера
| Показатели | Метод | Цель исследования | Больные, способные к самостоятельному передвижению | Больные, не способные к самостоятельному передвижению |
| Исследование силы | Мануальное исследование мышц, количественная миометрия | Мониторинг клинического течения, прогнозирование утраты функций; оценка эффективности лечения; выявление асимметрии мышечной силы | Определение силы нижних конечностей с помощью мануального исследования каждые 6 мес. | На ранних стадиях: определение силы верхних и нижних конечностей каждые 6 мес. На поздних стадиях: ценность данного исследования неясна |
| Объем движений | Гониометрия | Выявление развивающегося снижения мышечной экстензии и наличия суставных контрактур; необходимость дополнительного/альтернативного медикаментозного/хирургического вмешательства | Нижние конечности: бедренные, коленные, голеностопные суставы; илиотибиальный тракт; подколенные сухожилия, икроножная мышца | Нижние конечности: бедренные, коленные, голеностопные суставы; илиотибиальный тракт; подколенные сухожилия, икроножная мышца. Верхние конечности: локтевой сустав, запястье, длинные сгибатели пальцев |
| Тесты на время | Выполнение с учетом времени стандартизованных заданий для проверки различных функций | Легкие в практическом применении и информативные показатели повседневного функционального состояния; чувствительны к изменению состояния | Время прохождения 10 м, время выполнения приема Говерса, время подъема на 4 ступеньки, тест с 6-минутной ходьбой | Время, необходимое для одевания рубашки, может быть информативным показателем на ранних этапах данной стадии. Тесты на время не применимы на поздних этапах стадии |
| Повседневная деятельность | Оценка ограничений в повседневной деятельности дома, в школе и общественных местах | Выявление точек приложения помощи, адаптации и доступа к контролируемой окружающей среде | Частота падений, мониторинг количества шагов, навыки самообслуживания, письма и работы за компьютером. Дееспособность в школе и общественных местах | Навыки самообслуживания, письма и работы за компьютером, управление ручным и электрическим креслом-каталкой. |
| Шкалы двигательных функций | Оценка двигательных функций отдельных областей с получением объединенной оценки | Мониторинг прогрессирования болезни и эффекта лечения | Шкала оценки функций нижних конечностей Vignos, шкала оценки способности к самостоятельному передвижению North Star, шкала измерения двигательной функции | Шкала оценки функционального рейтинга верхних конечностей по Brooke, функциональная оценка по Egen Klassifikation, двигательные шкалы Hammersmith, шкала измерения двигательной функции |
Диагностический алгоритм
Диагностический алгоритм ПМД Дюшенна [3]

Диагностика (стационар)
ДИАГНОСТИКА НА СТАЦИОНАРНОМ УРОВНЕ
Диагностические критерии на стационарном уровне:
Жалобы пациента с верифицированным диагнозом «ПМД Дюшенна/
Беккера»:
· побочные эффекты гормональной терапии;
· прогрессирование двигательных нарушений;
· развитие кардиологических осложнений заболевания;
· развитие ортопедических осложнений заболевания;
· развитие дыхательных осложнений заболевания;
Анамнез:
· наличие в анамнезе клинически установленного ПМД Дюшенна/
Беккера;
· прием глюкокортикоидной терапии при ПМД Дюшенна;
· недифференцированное нервно-мышечное заболевание у пациента мужского пола в возрасте от 2 до 10 лет при высоком уровне трансаминаз и КФК крови, обнаруженном амбулаторно;
· невозможность проведения специфического обследования (МРТ мышц, генетическое исследование, биопсия мышц) на амбулаторно-поликлиническом уровне.
Физикальное обследование: см. амбулаторный уровень.
Лабораторные исследования:
· ОАК
· ОАМ
· биохимический анализ крови
· глюкоза крови
· электролиты
Инструментальные исследования:
· УЗИ внутренних органов;
· ЭКГ, ЭхоКГ;
· ЭНМГ;
· УЗИ мышц голени, бедра;
МРТ/МРС мышц:
· количественная и качественная оценка скелетных мышц;
· основные параметры: площадь поперечного сечения мышцы, % соотношение
фракции жира, признаки повреждения/воспаления.
Генетические исследования:
· методы: мультиплексная ПЦР, мультиплексная лигазная амплификация зонда,
амплификация при постоянных условиях/внутренний праймер, секвенирование гена;
· поиск мутаций гена дистрофина DMD в образце крови: делеции или
дупликации.
Биопсия мышц с последующим гистохимическим исследованием биоптата:
· открытая, инцизионная биопсия показана для проведения дифференциальной
диагностики ПМД Дюшенна и других мышечных дистрофий; пункционная, игольчатая биопсия – диагностика исключительно ПМД
Дюшенна;
· полное отсутствие дистрофина характерно для ПМД Дюшенна, частичное
отсутствие – для ПМД Беккера и других дистрофинопатий с доброкачественным фенотипом.
· дистрофические изменения, перимизиальное рубцевание, мышечные волокна
разного калибра, лимфоцитарная инфильтрация и патологическая регенерация.
NB! Биопсия мышц выполняется в зависимости от клинической ситуации, доступности проведения генетического исследования и технических возможностей клиники, где наблюдается пациент.
NB! Проведение генетического исследования после получения положительного результата по данным биопсии мышц является обязательным.
NB! Биопсию мышц не целесообразно проводить если диагноз «ПМД Дюшенна» был изначально установлен на основании генетического исследования.
Диагностический алгоритм: см.амбулаторный уровень.
Перечень основных диагностических мероприятий:
· ферменты крови (АЛТ, АСТ, КФК, ЛДГ);
· ЭНМГ;
· УЗИ мышц;
· МРТ/МРС мышц;
· генетическое исследование;
· биопсия.
Перечень дополнительных диагностических мероприятий:
· развернутый ОАК;
· развернутый биохимический анализ крови (общий билирубин и фракции, общий белок, тимоловая проба);
· глюкоза крови и мочи:
· ОАМ;
· электролиты (калий, кальций, натрий);
· 25-гидроксивитамин Д, ЩФ;
· ЭКГ, ЭхоКГ;
· спирометрия/спирография:
· денситометрия костей;
· УЗИ органов брюшной полости;
· МРТ/КТ позвоночника;
· рентгенография позвоночника в 2-х проекциях;
· рентгенография кисти.
Дифференциальный диагноз
Дифференциальный диагноз и обоснование дополнительных исследований
Основные заболевания для дифференциального диагноза ПМД Дюшенна/Беккера
| Тип нарушения | Диагноз | Основные схожие симптомы |
| Воспалительная миопатия | Полимиозит Миозит с включениями | Постепенно развивающаяся слабость мышц, повышение КФК |
| Врожденные миопатии | Немалиновая миопатия Болезнь центрального стержня и мультистержневая миопатии Центронуклеарная миопатия Миопатия с гиалиновыми тельцами Прочие врожденные миопатии | Мышечная слабость, гипотония при нормальном или умеренно повышенном уровне КФК, наличие скелетных нарушений |
| Метаболические миопатии | Гликогеноз II типа (болезнь Помпе) Гликогенозы IIIа, IV V и VII типов Болезнь МакАрдля (поздняя форма) Митохондриальные миопатии Жировые миопатии | Гипотония, слабость мышц, утомляемость, снижение устойчивости к нагрузкам, повышение КФК, кардиомиопатия, плотные на ощупь икроножные мышцы, возможен прием Говерса |
| Болезни мотонейрона | Спинальные мышечные атрофии, тип I, II и III Бульбоспинальная амиотрофия (болезнь Кеннеди) Боковой амиотрофический склероз | Слабость мышц, атрофия мышц, кардиомиопатия, наличие скелетных нарушений, возможно повышение КФК, респираторные нарушения |
| Болезни нервно- мышечной передачи | Миастения гравис Врожденные миастенические синдромы Синдром Ламберта-Итона | Слабость мышц, утомляемость, респираторные нарушения |
Лечение
Препараты (действующие вещества), применяющиеся при лечении
| Дефлазакорт (Deflazacort) |
| Колекальциферол (Kolekaltsiferol) |
| Левокарнитин (Levocarnitine) |
| Пиридоксин (Pyridoxine) |
| Преднизолон (Prednisolone) |
| Преднизон (Prednisone) |
| Тиамин (Thiamin) |
| Цианокобаламин (Cyanocobalamin) |
Лечение (амбулатория)
ЛЕЧЕНИЕ НА АМБУЛАТОРНОМ УРОВНЕ
Тактика лечения:
Немедикаментозное лечение:
· диета, обогащенная витаминами, микроэлементами, пищевые добавки,
содержащие кальций, витамины группы В, Д;
· адекватная физическая активность под контролем родителей, мед. персонала,
инструктора;
Медикаментозное лечение:
Терапия глюкокортикоидами – единственный из доступных методов медикаментозного лечения, который позволяет замедлить утрату мышечной силы и функций, уменьшить риск развития ортопедических осложнений и стабилизирует функциональное состояние легких и сердца.
· преднизон/преднизолон 0,75 мг/кг/сутки, утренний прием – препарат первой
линии при отсутствии таких факторов, как избыток веса и/или нарушение поведения;
· дефлазокорт 0,9 мг/кг/сутки, утренний прием – препарат первой линии при
наличии существующих проблем с весом и/или поведением.
NB! Необходимо принять во внимание возраст, функциональное состояние (улучшение, плато, угасание), уже существующие факторы риска, взаимоотношения врача с семьей;
NB! До начала приема ГК должен быть выполнен календарь иммунизации;
Алгоритм начала ГК терапии у детей с ПМД Дюшенна
| Возраст | Тактика лечения ГК в зависимости от функционального состояния |
| младше 2-х лет | • Развитие (типично): начинать лечение ГК не рекомендуется; • Плато (редко): тщательное наблюдение; • Угасание (не характерно): рассмотреть возможность другого диагноза или сопутствующего заболевания. |
| возраст 2-5 лет | • Развитие: начинать лечение ГК не рекомендуется; • Плато: рекомендуется начинать лечение ГК; • Угасание: настоятельно рекомендуется начинать лечение ГК. |
| возраст 6 лет и старше | • Развитие (редко): рассмотрите вероятность миодистрофии Беккера; • Плато: настоятельно рекомендуется начинать лечение ГК; • Угасание: настоятельно рекомендуется начинать лечение ГК. |
Метаболическая терапия – терапия, направленная на улучшение обменных процессов в скелетных мышцах, костной ткани, печени, миокарде; нормализацию белкового и жирового обмена, угнетает образование кетокислот, снижает лактат-ацидоз; для профилактики и устранения побочных эффектов гормональной терапии.
· витамины группы В;
· витамин Д3;
· левокарнитин;
· препараты кальция.
Перечень основных лекарственных средств:
· преднизон/преднизолон – таблетка 5 мг;
· дефлазакорт – таблетка 30 мг;
Профилактические мероприятия:
· медико-генетическое консультирование отягощенных семей – основное
профилактическое мероприятие;
· пренатальная диагностика плода у женщины-носительницы патологического
гена;
· профилактика серьезных осложнений заболевания;
· профилактика побочных эффектов гормональной терапии.
Лечение (скорая помощь)
ДИАГНОСТИКА И ЛЕЧЕНИЕ НА ЭТАПЕ СКОРОЙ НЕОТЛОЖНОЙ ПОМОЩИ
Диагностические мероприятия: нет.
Медикаментозное лечение:
Медикаментозное лечение, оказываемое на этапе скорой неотложной помощи (смотреть КП по соответствующим нозологиям):
· лечение острой сердечной недостаточности;
· лечение аритмии;
· лечение острой дыхательной недостаточности.
Лечение (стационар)
ЛЕЧЕНИЕ НА СТАЦИОНАРНОМ УРОВНЕ
Тактика лечения: см. амбулаторный уровень.
Немедикаментозное лечение: см. амбулаторный уровень.
Медикаментозное лечение: см. амбулаторный уровень.
Хирургическое вмешательство:
Вид операции:
· хирургическая коррекция суставов и связок при контрактурах нижних конечностей, патологических установках стоп;
· хирургическая коррекция и стабилизация позвоночника при сколиозе;
· внутренняя фиксация, шинирование, гипсование – при переломах костей;
· трахеотомия – при неэффективности неинвазивной вентиляции легких, аспирационном синдроме;
· гастростомия – при значительной потере веса, невозможности восполнения дефицита питательных веществ и воды пероральными средствами;
Другие виды лечения:
· поддержание функционального объема легких с помощью самонадувающихся ручных дыхательных мешков;
· методы ручного и механического облегчения откашливания;
· вентиляция легких в ночное и/или дневное время;
· вакцинация против пневмококковой инфекции и гриппа;
· терапия болевого синдрома: миорелаксанты, нестероидные противовоспалительные средства.
· психотерапия индивидуальная, семейная.
Показания для перевода в отделение интенсивной терапии и реанимации:
· острая дыхательная недостаточность;
· острая сердечная недостаточность;
· артериальная гипертензия, криз.
Индикаторы эффективности лечения:
· стабилизация общего состояния;
· улучшение самочувствия;
· нормализация лабораторных показателей;
· купирование симптомов дыхательной недостаточности;
· купирование симптомов сердечной недостаточности;
· удовлетворительная переносимость гормональной терапии.
Медицинская реабилитация
Для оказания данного вида помощи требуется участие специалистов в области нервно-мышечной патологии, физиотерапевта, реабилитолога и специалистов по ортопедической хирургии. Реабилитолог осуществляет мониторинг и применение программ реабилитации, адаптацию/настройку под индивидуальные потребности пациента в зависимости от стадии заболевания, реакции на терапию и переносимости. На амбулаторном этапе помощь оказывается специалистом каждые 4 месяца.
Методы физической реабилитации:
· активное растягивание;
· активное растягивание с посторонней помощью;
· пассивное растягивание;
· длительное вытяжение с использованием позиционирования, шинирования;
· ортезы (AFO – голень-стопа; KAFO – колено-голень-стопа);
· приспособления для вертикализации.
Хирургическое вмешательство при контрактурах нижних конечностей
Вспомогательные приспособления для компенсации функций и адаптации:
· ручные кресла-коляски;
· моторизированные кресла-коляски;
· автоматически регулируемые кровати;
Физические нагрузки: для предотвращения атрофии и вторичных осложнений пациенты с сохранной способностью к самостоятельному передвижению и пациенты на ранней стадии потери способности к самостоятельному передвижению должны регулярно заниматься укреплением мышц в субмаксимальном (легком) режиме под наблюдением инструкторов, мед. персонала, обученных родителей..
· плавание;
· восстанавливающие упражнения рекреационной направленности.
NB! Силовая тренировка и эксцентрические упражнения противопоказаны.
Междисциплинарные взаимодействия при оказании помощи больным ПМД Дюшенна [4,5]

Госпитализация
Показания для плановой госпитализации:
· проведение дифференциальной диагностики и установление окончательного диагноза;
· подбор оптимального режима и дозы глюкокортикоидов.
Показания для экстренной госпитализации:
· наличие серьезных осложнений со стороны сердечно-сосудистой, костно-суставной, дыхательной систем;
· наличие серьезных побочных эффектов гормональной терапии.
Информация
Источники и литература
Информация
Сокращения, используемые в протоколе
Список разработчиков протокола:
1) Лепесова Маржан Махмутовна – доктор медицинских наук, профессор, заведующая кафедрой детской неврологии с курсом медицинской генетики КазМУНО, независимый аккредитованный эксперт по детской неврологии, детский невролог высшей квалификационной категории, Президент ОО «Ассоциация детских неврологов Республики Казахстан», член международных профессиональных ассоциаций (ICNA, EPNS, AOCNA, EACD).
2) Мырзалиева Бахыткуль Джусупжановна – магистр медицинских наук, главный внештатный детский невролог УЗ г. Алматы, детский невролог высшей квалификационной категории, докторант Международного Казахско-Турецкого университета имени Х.А. Яссави, член ОО «Ассоциация детских неврологов Республики Казахстан» и международных профессиональных ассоциаций.
3) Тулеутаеву Райхан Есенжановна – кандидат медицинских наук, заведующая кафедрой фармакологии и доказательной медицины Государственного медицинского университета г. Семей, член «Ассоциации врачей терапевтического профиля».
Указание на отсутствие конфликта интересов: нет
Список рецензентов: Джаксыбаева Алтыншаш Хайруллаевна – доктор медицинских наук, руководитель отделения психоневрологии АО «ННЦМД», главный внештатный детский невролог МЗиСР РК, детский невролог высшей квалификационной категории.