Смит-Магенис синдром
OMIM 182290
Наша команда профессионалов ответит на ваши вопросы
Заболевание проявляется черепно-лицевыми аномалиями, характерным строением тела, особенностями развития и поведения. Среди черепно-лицевых аномалий выделяют брахицефалию, плоское, широкое лицо, гипоплазию средней части лица, широкую переносицу, выпуклый лоб, сросшиеся брови, приоткрытый рот с шатровидной верхней губой, нижнюю микрогнатию, со временем переходящую в прогнатию; низко посаженные уши и другие аномалии ушей. К особенностям строения тела относят: широкие короткие кисти, брахидактилию, тугоподвижность локтевых суставов, плоскостопие. Также при данном заболевании обнаруживают врожденные пороки сердца, пороки развития почек, аномалии головного мозга, аномалии глаз, косоглазие, смешанную тугоухость, сколиоз, низкий хриплый голос, низкую болевую чувствительность. В грудном возрасте выявляется мышечная гипотония.
У детей наблюдается задержка речевого развития (моторная речь страдает больше сенсорной), IQ колеблется от 20 до 78 (у большинства между 40 и 54). К особенностям поведения относят саморазрушительное поведение: ребенок бьется головой, кусает руки, выгрызает или выдергивает ногти и т.п. Также для данного заболевания характерны расстройства сна: раннее засыпание (около 8 часов вечера), прерывистый сон (1—3 пробуждения за ночь), очень раннее утреннее пробуждение (в 4—6 часов) и сонливость в течение дня. Этот патологический ритм сна и бодрствования вызывает раздражительность и вспышки гнева.
Дупликация участка в 3.7 м.н.п. 17-й хромосомы ассоциирована с синдромом Потоцки-Лупски (OMIM610883), который имеет некоторые схожие черты с синдромом Смит-Магенис, но более мягкое течение.
Синдром смита магениса что это такое
Синдром Смита-Магениса представлен очень характерным поведенческим фенотипом. Заболевание вызвано интерстициальной делецией хромосомы 17р11.2 (Greenberg et al., 1991, 1996).
Редкие случаи развития синдрома без данной делеции связаны с вновь возникшей мутацией гена RAI1 (Slager et al., 2003; Girirajan et al., 2005; Bi et al., 2006). Пациенты с данным синдромом отстают в общем развитии, обычно с умеренной задержкой умственного развития (хотя у некоторых пациентов задержка умственного развития выражена слабо).
В большинстве случаев отмечается брахицефалия, гипоплазия средней части лица, мальформации ушей и брахидактилия. Аномалии ЛОР-органов выявляются практически у всех пациентов, а аномалии глаз — у 85% пациентов; также возможны аномалии других внутренних органов (Greenberg et al., 1996).

У трех четвертей пациентов отмечается нарушение сна с инверсией циркадного ритма секреции мелатонина (De Leersnyder et al., 2001a). В большинстве случаев выявляются выраженные поведенческие отклонения с агрессивностью и самодеструктивным поведением (включая онихотилломанию и введение инородных предметов в различные физиологические отверстия).
Удары головой, кусание рук и другие стереотипные действия (необычные для других заболеваний), сжимание кистей или рук, иногда описываемое как «спазматическое сжимание верхней части тела» или «обнимание самого себя» (Finucane et al., 1994), являются характерными проявлениями данного синдрома. Такое поведение, являющееся основным клиническим признаком, часто возникает в знакомой обстановке, когда пациент расслаблен и находится в хорошем настроении; выявление такой особенности является основанием для проведения хромосомного анализа.
Данный признак очень важен, так как появление такого поведения в обстановке медицинского учреждения достаточно нетипично. Большинство пациентов нежны и отличаются положительным эмоциональным отношением несмотря на вспышки агрессии и самодеструктивного поведения.
У девяти пациентов с синдромом Смита-Магениса отмечалось положительное воздействие антагонистов бета-1-адренорецепторов на сон и поведение (De Leersnyder et al, 2001b).
Редактор: Искандер Милевски. Дата публикации: 6.12.2018
Синдром Смит-Магенис

Синдром Смит-Магенис — это редкое генетическое заболевание, которое возникает при микроповреждении короткого плеча 17-й хромосомы. Патология проявляется множественным врожденными пороками развития, среди которых преобладают аномалии лицевого скелета, а также интеллектуальными нарушениями, разнообразными поведенческими особенностями. Для диагностики синдрома Смит-Магенис назначается молекулярно-генетическое тестирование, неврологический обследование, методы инструментальной визуализации. Больным требуется помощь логопедов, реабилитологов, коррекционных педагогов, по показаниям проводится лечение соматических осложнений.
МКБ-10

Общие сведения
Синдром назван в честь американских врачей ‒ клинициста Энн Смит и цитогенетика Эллен Магенис, которые в 1980 г. впервые описали характерный симптомокомплекс у группы детей. Ранее считалось, что заболевание встречается с частотой 1:25000, но с развитием молекулярно-генетических методов диагностики подтвердилась более высокая распространенность — 1:15000. Чаще всего патология наблюдается у детей с задержкой психического развития — 1 случай на 569 пациентов.
Причины
Заболевание возникает вследствие спонтанной хромосомной аномалии — делеции 17 хромосомы в локусе 17p11.2, которая предполагает потерю до 4 млн. нуклеотидных пар. До 25% клинических случаев синдрома Смит-Магенис составляют атипичные микроделеции, которые выражаются в утрате меньшего участка короткого плеча хромосомы. Болезнь носит спорадический характер, пока ученым не удалось установить типичные закономерности и провоцирующие факторы.
Патогенез
Развитие характерной для синдрома симптоматики прежде всего связывают с нарушением гена RAI1, который является индуктором ретиноловой кислоты. Ученые обсуждают его участие в дифференцировке тканей, что объясняет формирование множественных врожденных пороков. Также RAI1 предположительно используется при дифференцировке нейронов. Некоторые психические проявления синдрома Смит-Магенис вызваны дефектом синтеза и деградации меланина.
Симптомы
Дети имеют типичные особенности внешности, которые обнаруживаются уже в младенческом возрасте. Обычно у них широкая квадратная форма головы, выпуклый лоб, глубоко посаженные глаза с монголоидным разрезом. Также для синдрома характерно недоразвитие средней зоны лица, короткий нос с вывернутыми ноздрями, шатровидная верхняя губа при относительно маленькой нижней челюсти. У детей непропорционально короткие руки и ноги, с раннего возраста отмечается задержка роста.
Специфическим диагностическим признаком синдрома Смит-Магенис являются нарушения сна. На первом году жизни наблюдается патологическая сонливость, такие дети много спят днем, их приходится будить для очередного кормления. Со временем нарушаются циркадные ритмы, преобладает поздний отход ко сну с частыми ночными пробуждениям. С возрастом пациент все больше спит днем, не может заснуть ночью.
Психоневрологические изменения включают умственную отсталость умеренной степени тяжести, возникающую практически у всех больных. Характерна задержка психоречевого развития, затруднения при произношении слов, дефицит внимания с гиперактивностью. До 30% пациентов имеют неврологические нарушения: расстройства координации движений, периферическую нейропатию, судорожные приступы.
Болезнь Смит-Магенис сопровождается специфическим поведенческим профилем. Дети страдают стереотипным поведением, проявляют дезадаптивные черты личности — вспышки гнева, непослушание, постоянное требование внимания к себе со стороны взрослых. Некоторые больные склонны к самоповреждению: кусают, щиплют или бьют себя, вводят инородные предметы в физиологические отверстия тела.
Осложнения
Большинство детей имеют мышечную гипотонию с рождения, вследствие чего возникают трудности вскармливания, нарушения глотания, отказ от пищи. В 80% случаев синдром Смит-Магенис ассоциирован с офтальмологическими проблемами: дисплазией радужки, микрокорнеа, пятнами Брушфильда. Более 60% больных страдают хроническими отитами, кондуктивной или нейросенсорной тугоухостью. У половины пациентов диагностируется гиперхолестеринемия.
Пороки сердца встречаются в 25% случаев синдрома. Наиболее распространены дефекты межпредсердной и межжелудочковой перегородок, стеноз трикуспидального клапана, тетрада Фалло. До 15% детей страдают от аномалий мочевыводящих путей, которые включают унилатеральную агенезию почек, удвоение мочеточников, увеличение линейных размеров органов.
Диагностика
Обследование осуществляет мультидисциплинарная команда специалистов с участием педиатра, детского невролога, психиатра, генетика. Синдром Смит-Магенис имеет патогномоничные фенотипические и поведенческие признаки, при выявлении которых устанавливается предварительный диагноз. Для верификации патологии используются следующие методы:
Лечение синдрома Смит-Магенис
Показано ежегодное комплексное обследование состояния здоровья для контроля динамики нарушений. Больному подбирается индивидуальная программа симптоматической терапии. Медикаментозные средства назначаются редко, в основном для коррекции бессонницы, а психотропные препараты не используются из-за риска усугубления психических проблем. Лечение включает следующие направления:
При осложненном течении синдрома схема лечения расширяется. Неврологические расстройства требуют применения физиотерапии, упражнений ЛФК, противоэпилептических препаратов. Для нормализации зрения некоторым необходима оптическая коррекция, исправление косоглазия. Хирургическое лечение пороков сердца осуществляется в соответствии с выявленной аномалией.
Прогноз и профилактика
При своевременном начале комплексной терапии синдрома Смит-Магенис удается значительно развить моторные и речевые навыки пациента, адаптировать его к жизни в социуме. Прогноз относительно благоприятный при пожизненной реабилитации, социально-психологической поддержке. Специфические меры профилактики синдрома не разработаны, что обусловлено его спорадическим характером.
Синдром Смита-Магениса
Внимание! Информация носит ознакомительный характер и не предназначена для постановки диагноза и назначения лечения. Всегда консультируйтесь с профильным врачом!
Синдром Смита-Магениса (ССМ) — сложное нарушение развития, которое поражает несколько систем органов тела. Расстройство характеризуется набором аномалий, которые присутствуют при рождении (врожденные), а также поведенческими и когнитивными проблемами. Общие симптомы включают характерные черты лица, пороки развития скелета, различные степени умственной отсталости, задержку речи и моторики, нарушения сна, а также самоповреждающее поведение, направленное на привлечение внимания. Конкретные симптомы, присутствующие у каждого пациента, могут сильно отличаться от одного человека к другому. Примерно 90% случаев ССМ вызваны отсутствием или удалением части хромосомы (моносомная). Эта удалением части в хромосоме 17p11.2 включает ген, который, как полагают, играет главную роль в развитии заболевания. В остальных случаях удаленный материал на 17 хромосоме отсутствует; эти случаи вызваны мутациями в гене RAI1. Другие гены в удаленном сегменте также могут играть роль в вариабельных признаках синдрома Смита-Магениса, но не совсем понятно, насколько важную роль они играют в развитии ССМ.
Синдром Смита-Магениса впервые был описан в медицинской литературе в 1982 году генетическим консультантом Энном Смитом и ее коллегами. В 1986 году Смит и доктор Р. Эллен Магенис идентифицировали 9 пациентов с расстройством, что позволило более точно определить характер синдрома. С тех пор было выявлено множество дополнительных случаев, что позволило врачам/клиницистам лучше понять это сложное расстройство развития.
Признаки и симптомы
Основными признаками синдрома Смита-Магениса являются легкая и умеренная умственная отсталость, задержка речи и моторики, отличительные черты лица, нарушения сна, скелетные и зубные аномалии, а также поведенческие проблемы.
Черты лица у пациентов с ССМ могут быть незаметными в раннем детстве, но обычно становятся более очевидными с возрастом и включают:
Несмотря на то, что людей с ССМ часто характеризуются как ласковые, обаятельные личности, большинство из них также имеют проблемы с поведением. Они включают:
Дополнительные симптомы ССМ включают:
Причины и факторы риска
Примерно у 90% больных часть короткого плеча (p) хромосомы 17 (17q11.2) отсутствует, что называется моносомия. Хромосомы, присутствующие в ядре клеток человека, несут генетическую информацию каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин одна X и одна Y хромосома, а у женщин две X хромосомы. Каждая хромосома имеет короткое плечо, обозначенное буквой «p», и длинное плечо (руку), обозначенное буквой «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 17p11.2» относится к полосе 11.2 на коротком плече хромосомы 17. Пронумерованные полосы указывают расположение тысяч генов, присутствующих на каждой хромосоме.
У пациентов с синдромом Смита-Магениса удаленная часть (моносомия) хромосомы 17 включает ген RAI1. Гены предоставляют инструкции по созданию белков, которые играют решающую роль во многих функциях организма. Когда ген отсутствует из-за аномалии моносомной хромосомы, белковый продукт этого гена уменьшается. В зависимости от функций конкретного белка это может повлиять на многие системы органов тела, включая мозг.
Точная причина хромосомного изменения при ССМ неизвестна. Медицинская литература указывает на то, что практически все задокументированные случаи связаны со спонтанными (новыми) генетическими изменениями, которые происходят по неизвестным причинам.
В редких случаях ССМ является результатом ошибки во время очень раннего эмбрионального развития из-за хромосомно-сбалансированной транслокации у одного из родителей. Транслокация сбалансирована, если части двух или более хромосом отламываются и меняются местами, создавая измененный, но сбалансированный набор хромосом. Если хромосомная перестройка сбалансирована, она обычно безвредна для носителя. Однако они могут быть связаны с более высоким риском аномального развития хромосом у потомков носителя. В этих случаях на клинические особенности детей может влиять дополнительный дисбаланс других хромосом, кроме 17. Хромосомное исследование может определить, есть ли у родителя сбалансированная транслокация. У родителей с ребенком с ССМ и нормальным хромосомным анализом риск рецидива при будущей беременности ниже 1%.
Остальные 10% случаев синдрома Смита-Магениса вызваны мутациями в гене RAI1, что приводит к недостаточному уровню функциональных копий белкового продукта, обычно продуцируемого этим геном.
В двух семьях, о которых говорится в медицинской литературе, ССМ произошел из-за мозаицизма зародышевой линии. При мозаицизме зародышевой линии некоторые из родительских репродуктивных (зародышевых) клеток несут мутацию RAI1 гена или делецию 17p хромосомы, в то время как другие половые клетки этого не делают (мозаицизм). Кроме того, другие клетки родителя также не имеют этих хромосомных аномалий; следовательно, это не влияет на родителей. Однако в результате один или несколько детей родителей могут унаследовать половые клетки с хромосомной аномалией, что приведет к развитию ССМ. Мозаицизм зародышевой линии подозревается, если у явно здоровых родителей есть более одного ребенка с этим заболеванием. Вероятность того, что родитель передаст ребенку мозаичную хромосомную аномалию зародышевой линии, зависит от процента половых клеток родителя, которые имеют аномалию, по сравнению с процентом, который не имеет. Исследований на мутации зародышевой линии или хромосомные аномалии до беременности не проводится.
У ребенка, рожденного от человека с синдромом Смита-Магениса, теоретический риск 50% унаследовать делецию или мутацию RAI1, которая вызывает заболевание. Рождаемость в ССМ в целом до конца не изучена; однако в медицинской литературе есть как минимум одно сообщение о матери с ССМ, имеющей ребенка с ССМ.
Затронутые группы населения
Синдром Смита-Магениса поражает мужчин и женщин в равном количестве. Заболевание обнаруживается у 1 из 15 000–25 000 человек среди населения. Однако случаи могут остаться недиагностированными или ошибочно диагностированными, что затрудняет определение истинной частоты расстройства среди населения в целом. ССМ обнаруживался во всем мире и среди всех этнических групп.
Близкие по симптомам расстройства
Симптомы следующих заболеваний могут быть похожи на симптомы синдрома Смита-Магениса. Сравнения могут быть полезны для дифференциальной диагностики.
Существует множество различных хромосомных нарушений, признаки и симптомы которых аналогичны тем, которые наблюдаются при ССМ. К таким расстройствам относятся:
Пациентам с ССМ часто сначала ставится психиатрический диагноз, включающий обсессивно-компульсивное расстройство, общие расстройства психологического развития или синдром дефицита внимания с гиперактивностью. Некоторым детям с ССМ изначально ставится диагноз расстройства аутистического спектра.
Диагностика
Диагноз синдрома Смита-Магениса основывается на выявлении характерных симптомов, подробном анамнезе пациента и его семьи, тщательной клинической оценке и различных специализированных генетических тестах. Диагноз ССМ подтверждается при идентификации делеции 17p11.2 (цитогенетический анализ или ДНК-микрочип) или мутации гена RAI1.
— Клиническое обследование.
В прошлом для диагностики синдрома Смита-Магениса использовалось специальное хромосомное исследование, известное как анализ G-диапазона, которое демонстрирует отсутствие (удаленный) материала на хромосоме 17p. Хромосомы можно получить из образца крови. Во время этого теста хромосомы окрашиваются, чтобы их было легче увидеть, а затем исследуются под микроскопом, где можно обнаружить отсутствующий сегмент хромосомы 17p (кариотипирование). Для определения точной точки останова может потребоваться более чувствительный тест, известный как флуоресцентная гибридизация in situ (FISH). Во время исследования FISH зонды, отмеченные определенным цветом флуоресцентного красителя, прикрепляются к определенной хромосоме, что позволяет исследователям лучше видеть эту конкретную область хромосомы.
Также можно использовать более новый метод, известный как анализ хромосомных микрочипов. Во время этого анализа ДНК человека сравнивается с ДНК человека без хромосомной аномалии («контрольный» человек). Хромосомная аномалия отмечается при обнаружении различий между образцами ДНК. Хромосомный микроматричный анализ позволяет обнаруживать очень небольшие изменения (отсутствующие или дублированные сегменты) или изменения.
Молекулярно-генетическое тестирование может подтвердить диагноз у лиц, подозреваемых в наличии ССМ из-за мутации гена RAI1. Молекулярно-генетическое тестирование может обнаруживать мутации в гене RAI1, которые, как известно, вызывают ССМ в определенных случаях, но доступны только в качестве диагностической услуги в специализированных лабораториях.
Стандартные методы лечения
Лечение может потребовать скоординированных усилий команды специалистов. Педиатрам, хирургам, кардиологам, стоматологам, логопедам, аудиологам, офтальмологам, психологам и другим медицинским работникам может потребоваться систематическое и всестороннее планирование и влияние на лечение ребенка. Генетическое консультирование полезно для пострадавших и их семей. Психосоциальная поддержка также важна для всей семьи.
Лечение симптоматическое и поддерживающее. Раннее вмешательство важно для того, чтобы затронутые дети раскрыли свой потенциал. Услуги, которые могут быть полезны, включают специальное лечебное обучение, речевую/языковую терапию, физиотерапию, трудотерапию и терапию сенсорной интеграции, при которой предпринимаются определенные сенсорные действия, чтобы помочь регулировать реакцию ребенка на сенсорные стимулы. При необходимости могут быть рекомендованы дополнительные медицинские, социальные и профессиональные услуги.
Некоторые лекарства используются для лечения поведенческих проблем, таких как дефицит внимания или гиперактивность. Для лечения расстройств сна, потенциально связанных с синдромом Смита-Магениса, также используются определенные лекарства. Добавки мелатонина для нормализации уровня мелатонина, принимаемые перед сном, показали свою пользу в отдельных отчетах. Использование B-блокатора ацебутолола утром для ингибирования/подавления дневной секреции мелатонина показало некоторую пользу в одном французском исследовании.
Проблемы с кормлением требуют выявления и соответствующей терапии. Дополнительное лечение следует стандартным рекомендациям для конкретного симптома. Например, противосудорожные препараты (противоэпилептические препараты) могут использоваться для лечения эпилепсии.
Прогноз
Из-за очень изменчивого характера синдрома невозможно сделать общий прогноз для отдельных случаев. Некоторые пострадавшие смогли найти работу и даже жить полунезависимо при поддержке семьи и друзей. Однако другие нуждаются в постоянном уходе, и им, возможно, придется жить с семьей или в приюте. Как указано выше, родители должны поговорить с врачом и медицинской командой о конкретном случае их ребенка и общем прогнозе.

Высшее образование (Кардиология). Врач-кардиолог, терапевт, врач функциональной диагностики. Хорошо разбираюсь в диагностике и терапии заболеваний дыхательной системы, желудочно-кишечного тракта и сердечно-сосудистой системы. Закончила академию (очно), за плечами большой опыт работ.
Специальность: Кардиолог, Терапевт, Врач функциональной диагностики.
Синдром смита магениса что это такое
Синдром Смита-Лемли-Опитца недавно стал объектом пристального внимания неврологов и психиатров из-за отчетливой взаимосвязи с аутистическими проявлениями. Возможно, данное заболевание имеет генетическую природу и тесно связано с аутистическими симптомами (см. ниже).
Синдром Смита-Лемли-Опитца (ССЛО) регистрируется с частотой около 1 на 70000 новорожденных. Характерна высокая перинатальная смертность и высокая частота мертворождений. Среди выживших детей не менее чем у 80% отмечаются тяжелые отклонения, а у оставшихся 20% изменения фенотипа выражены умеренно.
Синдром Смита-Лемли-Опитца (ССЛО) до сих пор диагностируется на основании типичных клинических признаков. Заболевание вызвано врожденным нарушением постскваленового синтеза холестерина. Нарушение синтеза холестерина при ССЛО вызвано наследственной мутацией гена 3-b-гидроксистерол-d-7 редуктазы (DHCR7). Повреждение гена DHCR7 приводит к нарушению синтеза холестерина и десмостерола, приводящее к повышению уровня 7DHC/8DHC, характерному снижению уровня холестерина и, что особенно важно, дисморфическому развитию.
Открытие синдрома Смита-Лемли-Опитца вызвало новые вопросы о роли путей биосинтеза холестерина в развитии человека. В настоящее время у 250 пациентов с синдромом Смита-Лемли-Опитца идентифицирована 121 мутация; разнообразие мутаций обеспечивает различную клиническую выраженность. Для воспроизведения некоторых аномалий развития, характерных для ССЛО, и выяснения патогенеза заболевания, было создано две генетических модели синдрома на мышах.
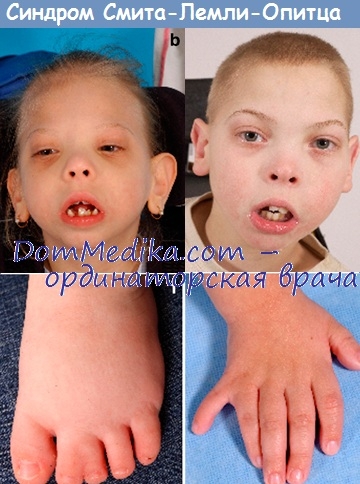
Наиболее характерными проявлениями синдрома наряду с задержкой умственного развития, аутизмом, другими поведенческими нарушениями и гипотонией является необычное строение лица с высоким плоским лбом, вывернутыми вперед ноздрями и микрогнатией, а также аномалии гениталий у мальчиков, включая крипторхизм и (или) гипоспадию, а в крайних случаях—неразвитые мужские гениталии, несмотря на нормальный кариотип XY (Scarbrough et al., 1986). Другие проявления включают аномальное строение ушных раковин, птоз век, эпикант, поперечное возвышение ладони и различные аномалии конечностей и внутренних органов (Opitz et al., 1994).
У некоторых пациентов отмечаются припадки, достаточно часто встречается отсутствие аппетита. В течение первого года жизни смертность высокая. Аномалии головного мозга могут включать микроцефалию, гипоплазию лобных долей, аномальное строение извилин и гипоплазию мозжечка. Случаи наиболее выраженного поражения иногда выделяются как ССЛО 2 типа (Curry et al., 1987).
Синдром наследуется аутосомно-рецессивным путем. Явное повышение частоты заболевания у мужчин, вероятно, связано с более легкой диагностикой на основании очевидных аномалий половых органов.
Концентрация холестерина в плазме очень низкая, в то время как содержание 7-гидрохолестерола повышено, что предполагает дефект фермента, приводящего к разрыву двойной связи гидрохолестерола (Tint et al., 1994). Выживаемость строго коррелирует с более высоким уровнем холестерина плазмы (Tint et al., 1995). Возможна пренатальная диагностика путем определения содержания 7-дегидрохолестерола в амниотической жидкости (Dallaire et al., 1995). Потребление большого количества холестерина может нормализовать его уровень в крови, но необязательно будет клинически эффективно (Ullrich et al., 1996).
Нарушения аутистического спектра часто регистрируются у пациентов с синдромом Смита-Лемли-Опитца (Gillberg и Coleman, 2000; Tierney et al., 2001; Sikora et al, 2006). Данные результаты и сообщение о низком уровне холестерина при различных случаях семейного аутизма позволяют предположить, что стериновый обмен может иметь важное значение для подгруппы пациентов с поведенческим синдромом аутизма и что хотя бы теоретически возможно «лечение» холестерином в случаях аутизма с низким уровнем холестерина.
Редактор: Искандер Милевски. Дата публикации: 6.12.2018