Синдром Паллистера-Киллиана
Оглавление
история
В 1977 году американский педиатр и генетик Филип Д. Паллистер впервые заподозрил этот синдром у двух взрослых. Независимо от этого, австрийский врач Вольфганг Киллиан и биолог-человек Мария Тешлер-Никола в 1981 году провели одинаковые наблюдения в общей сложности над четырьмя людьми.
генетика
частота
Точная частота синдрома Паллистера-Киллиана неизвестна. Возникает спорадически. То есть вероятность рецидива при последующих беременностях в семье не увеличивается. Говорят о « новой мутации ». В 2010 году в Германии и соседних странах было известно 38 детей с синдромом Паллистера-Киллиана в возрасте от 0 до 22 лет. Предполагается, что есть и другие пораженные дети, подростки или взрослые.
беременность
Симптомы
Наиболее частые симптомы синдрома Паллистера-Киллиана:
прогноз
Поскольку синдром Паллистера-Киллиана представляет собой мозаику хромосом, то есть избыток изохромосомы 12p присутствует не во всех клетках организма, невозможно сделать точных заявлений о доступном потенциале развития после хромосомной диагностики. Предполагается прямая связь между долей клеток с тетрасомией 12р в организме ребенка и тяжестью синдрома. Однако изучение отдельных тканей не позволяет сделать выводы о распределении двух клеточных линий по всему телу.
Диагностика
Диагноз синдрома Паллистер-Killian может быть затруднено. Обычный хромосомный анализ клеток крови обычно показывает нормальный набор хромосом. Линия клеток тетрасом в основном обнаруживается в культивируемых клетках кожи. Для этого у детей необходимо снять часть кожи ( биопсия кожи ). Теперь также возможна диагностика с помощью флуоресцентных зондов на клетках слизистой оболочки полости рта. В пренатальной хромосомной диагностике существующая тетрасомия не всегда может оказаться мозаикой 12p. Антенатальный диагноз синдрома Паллистера-Киллиана зависит от того, есть ли в исследуемом образце ткани тетрасомные клетки. Ребенок с синдромом Паллистера-Киллиана может появиться на свет даже после незаметного результата хромосомного исследования клеток околоплодных вод ( амниоцентеза ).
Синдром Паллистера-Киллиама Симптомы, причины, лечение
Синдром Паллистера-Киллиана (SPK), также известный под именем тетрасомия 12, это редкое заболевание генетического происхождения, характеризующееся широким спектром участия многих органов.

Кроме того, могут возникать и другие виды медицинских осложнений, связанные с пороками развития в различных системах организма или судорожными эпизодами (Толедо-Браво-де-ла-Лагуна и др., 2014)..
Этиологическое происхождение этого заболевания связано с генетическим изменением, распространенным в мозаике. В частности, это связано с наличием дополнительной хромосомы 12 в некоторых клетках организма (Понимание хромосомных нарушений, 2016).
Диагноз синдрома Паллистера-Киллиама может быть поставлен как на дородовой, так и на постнатальной стадии. Основной целью является выявление клинических характеристик и использование подтверждающего генетического исследования (Мендес, Родригес, Болуарте, Картолин, Вальдес и Матеус, 2013).
Этот синдром имеет высокий уровень смертности (Рамирес Ферандес, Гарсия Кавасос, Санчес Мартинес, 2007). Тем не менее, медицинский фармакологический подход и реабилитационное лечение могут обеспечить важные преимущества в качестве жизни и клиническом статусе пострадавших (Méndez et al., 2013).
Характеристика синдрома Паллистера-Киллиама
Некоторые учреждения, такие как Genectis Home Reference (2016), классифицируют эту патологию в рамках так называемых нарушений развития.
На общем уровне нарушения развития или нарушения развития в их международном термине, как правило, относятся к широкому набору физических и когнитивных изменений и аномалий. Все это приводит к отклонению или значительной задержке развития относительно нормальных или ожидаемых паттернов (Национальный институт неврологических расстройств и инсульт, 2015).
При синдроме Pallister-Killiam выявлено широкое поражение различных систем организма и организмов (Genectis Home Reference, 2016)
Он в основном характеризуется умственной отсталостью, мышечной гипотонией, развитием отличительных черт лица, изменением пигментации кожи или ростом волос, среди других врожденных изменений (Genectis Home Reference, 2016).
Кроме того, синдром Pallister-Kiliam является редким врожденным заболеванием (Turleau, 2009), которое может получить большое количество имен в медицинской литературе (Национальная организация по редким заболеваниям, 2016):
Это заболевание было впервые описано Паллистером в 1977 году (Толедо-Браво-де-ла-Лагуна и др., 2014).
В первых публикациях указывалось на два случая взрослых пациентов, течение которых характеризовалось различными признаками: судороги, мышечная гипотония, интеллектуальный дефицит, костно-мышечные и органические пороки развития, грубая конфигурация лица и изменения цвета кожи (Méndez et al. al., 2013).
Параллельно Teschler-Nicola и Killiam в 1981 году описали эту же клиническую картину у трехлетней девочки (Méndez et al., 2013).
Поэтому в первых клинических отчетах была сделана общая ссылка на медицинское состояние, характеризующееся сочетанием судорог, умственной отсталости и характерного физического фенотипа (Толедо-Браво-де-ла-Лагуна и др., 2014)..
Кроме того, уже в 1985 году Гильгенкрац смог выявить в первом случае на этапе беременности, что часто встречается в настоящее время благодаря современным методам диагностики (Méndez et al., 2013).
статистика
Показатели распространенности синдрома Паллистера-Киллиама точно не известны. Было поставлено не так много точных диагнозов, и большинство из них не были опубликованы в медицинской литературе (Понимание хромосомных расстройств, 2016).
Таким образом, все авторы и учреждения определяют этот синдром как редкую или редкую генетическую патологию в общей популяции (EuRed, 2016).
Около 15 лет назад синдром Паллистера-Киллиама был выявлен только в 100 случаях во всем мире. В настоящее время эта цифра превысила 200 пострадавших (Понимание хромосомных расстройств, 2016).
Эпидемиологические исследования позволили оценить частоту этого заболевания примерно в 5,1 случая на миллион новорожденных детей (Понимание хромосомных расстройств, 2016), хотя такие авторы, как Толедо-Браво-де-ла-Лагуна и сотрудники (2014), считают, что 25000.
Более высокой распространенности, связанной с социально-демографическими характеристиками пострадавших, выявлено не было. Синдром Паллистера-Киллиана может проявляться в любой половой, технической и / или расовой группе..
Признаки и симптомы
В клиническом течении синдрома Паллистера-Киллиана могут быть выявлены самые разнообразные признаки и симптомы. Все они связаны с аномалиями черепно-лицевой и / или скелетных мышц и когнитивными изменениями.
Настройки лица
Развитие черепно-лицевых пороков развития от фазы беременности к постнатальному и младенческому росту является одним из наиболее характерных медицинских признаков синдрома Паллистер-Киллиама.
Наиболее распространенные признаки и симптомы включают нарушения в различных черепных и лицевых структурах, которые приводят к грубому и нетипичному внешнему виду (Toledo-Bravo de la Laguna et al., 2014; Понимание хромосомных нарушений, 2016):
Костно-мышечные пороки развития
Несмотря на то, что изменения являются менее значимыми, чем изменения лица, очень часто можно наблюдать несколько скелетно-мышечных нарушений у пациентов, страдающих синдромом Паллистера (Понимание хромосомных расстройств, 2016):
Мышечная гипотония и психомоторная отсталость
Нарушения, связанные с мышечной структурой и подвижностью, являются еще одним из основных клинических признаков синдрома Паллистера-Киллиана (Понимание хромосомных расстройств, 2016):
Мышечная гипотония относится к выявлению аномально сниженного мышечного тонуса или напряжения. На визуальном уровне вялость и лабильность могут наблюдаться в разных группах мышц, особенно в конечностях..
Таким образом, мышечная и скелетная патология вызовет значительную задержку в приобретении различных двигательных навыков, как у новорожденных, так и у детей..
Хотя периоды разработки варьируются среди затронутых, наиболее распространенный календарь включает в себя следующие этапы:
Неврологические изменения
Другие аномалии
Хотя они встречаются реже, могут возникать и другие виды медицинских осложнений (Национальная организация по редким заболеваниям, 2016 г.) (Толедо-Браво-де-ла-Лагуна и др., 2014 г.):
причины
Происхождение синдрома Pallister-Killian связано с генетической аномалией в мозаике на хромосоме 12. Он влияет только на генетический материал некоторых клеток организма (Inage et al., 2010).
Хромосомы являются частью ядра всех клеток, найденных в организме человека. Они состоят из широкого спектра биохимических компонентов и содержат генетическую информацию каждого человека (Национальная организация по редким заболеваниям, 2016 г.).
У людей есть 46 различных хромосом, организованных парами и пронумерованных от 1 до 23. Кроме того, на индивидуальном уровне каждая хромосома имеет участок или короткое плечо, называемое «p», и еще одно длинное, называемое «q» (Национальная организация по редким расстройствам, 2016).
Аномалия влияет на хромосому 12 и приводит к наличию хромосомы с аномальной структурой, называемой изокромосома (Genetics Home Reference, 2016).
Таким образом, эта хромосома имеет тенденцию иметь два коротких плеча вместо одного из каждой конфигурации p (короткий) и длинный (q) (Genetics Home Reference, 2016).
Как следствие, наличие дополнительного и / или аномального генетического материала изменит нормальный и эффективный ход физического и когнитивного развития больного человека, приводя к клиническим характеристикам синдрома Паллистер-Киллиана (Genetics Home Reference, 2016).
диагностика
Синдром Pallister-Killian может быть идентифицирован во время беременности или в постнатальном периоде, основываясь на клинических характеристиках и результатах различных лабораторных тестов (Turleau, 2009).
Во время беременности чаще всего используются ультразвуковые исследования, амниоцентез или ворсин хориона (Turleau, 2009)..
В этом смысле анализ генетического материала зародыша может предложить нам подтверждение этой патологии путем выявления совместимых аномалий (Turleau, 2009)..
С другой стороны, если диагноз ставится после рождения, он имеет основополагающее значение (Понимание хромосомных расстройств, 2016):
лечение
Для лечения людей с синдромом Паллистера-Киллиана не было разработано никаких специальных методов лечения (Национальная организация по редким заболеваниям, 2016).
Синдром Pallister-Killian обычно ассоциируется с плохим прогнозом и высоким уровнем смертности (Рамирес Ферандес, Гарсия Кавасос, Санчес Мартинес, 2007).
Однако реабилитационное лечение, специальное образование и трудотерапия могут дать хороший функциональный прогноз и повысить качество жизни пострадавших..
Например, Мендес и его команда (2013) описывают случай реабилитационного лечения, который характеризуется:
Синдром Паллистера-Киллиана — это крайне редкое генетическое заболевание, которое возникает при тетрасомии по 12-й хромосоме. Его развитие связывают с Х-рецессивным типом наследования. Состояние проявляется множественными патологиями перинатального периода, тяжелыми формами умственной отсталости, разнообразными врожденными аномалиями внутренних органов и стигмами дизэмбриогенеза. Диагностика синдрома Паллистера включает генетическое тестирование, инструментальную визуализацию (УЗИ, КТ, МРТ), неврологическое обследование. Пациентам назначается поддерживающее лечение, комплексная программа реабилитации, регулярное диспансерное наблюдение.
МКБ-10
Общие сведения
Синдром Паллистера (Паллистера-Киллиана) имеет синонимичные названия тетрасомия 12р, синдром мозаичной изохромосомы 12р. Он встречается крайне редко, в медицинской литературе есть данные о 30 случаях болезни, причем 6 пациентов проживают в Российской Федерации. Патология впервые описана в 1977 г. ученым Ф. Паллистером. В 1981 г. М. Теслер-Никола и В. Киллиан независимо друг от друга более подробно охарактеризовали клиническую картину синдрома. Первый пренатально диагностированный случай заболевания датируется 1985 г.
Причины
Болезнь возникает вследствие редкого мозаичного хромосомного расстройства, при котором в генетическом материале клетки образуются 4 копии короткого плеча 12 хромосомы вместо двух, которые должны присутствовать в норме. Эти дополнительные копии, как правило, расположены в одной из хромосом (изохромосома). Предполагается, что у синдрома Паллистера Х-сцепленный рецессивный тип наследования, но данных для подтверждения этой гипотезы недостаточно.
Патогенез
Заболевание появляется в результате генных нарушений, которые вызваны присутствием мозаичного клона mos 47,+ i(12)(p10)/46. Мутация формируется при нерасхождении сестринских хроматид в фазе мейоза II при образовании женских половых клеток. После оплодотворения такой яйцеклетки наблюдается постзиготическое ошибочное деление центромеры и частичная ее потеря при последующих митозах, поэтому только часть клеток тела ребенка имеют тетрасомию 12р.
Симптомы
Синдром Паллистера — тяжелая болезнь, которая может завершаться внутриутробной гибелью плода. В остальных случаях в анамнезе определяются патологии течения беременности в виде раннего токсикоза, угрозы прерывания на поздних сроках, задержки внутриутробного развития плода. У новорожденных отмечаются неврологические осложнения вследствие гипоксически-ишемической энцефалопатии, проблемы с самостоятельным дыханием, средняя или низкая оценка по шкале Апгар.
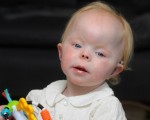
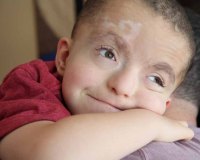
Больные с синдромом Паллистера имеют специфические особенности лицевого скелета: выступающий лоб с завышенной линией роста волос, низко посаженные деформированные уши, широкое расстояние между глазами. Также встречаются маленький вздернутый нос, вывернутые ноздри, большое расстояние от нижнего края ноздрей до верхней губы. На коже тела выявляются очаги депигментации.
К характерным особенностям синдрома относят диспропорциональное развитие конечностей с укорочением их проксимальных отделов, что сопровождается контрактурами суставов, стойким ограничением объема активных движений. Часто беспокоят проблемы со слухом и зрением. Для синдрома Паллистера-Киллиана типичны пороки сердца (дефект межжелудочковой перегородки, коарктация аорты, открытый Боталлов проток), диафрагмальная грыжа.
При тетрасомии 12р отмечается задержка психомоторного развития, умственная отсталость. Дети поздно начинают переворачиваться, сидеть и ползать, зачастую они не говорят даже в возрасте 1-3 лет, а только произносят отдельные звуки (гуление) или слоги. Наблюдается отставание в росте и физическом развитии, пациенты плохо набирают массу тела, имеют астеническое телосложение. Неврологические нарушения проявляются эпилепсией, мышечной гипотонией, тремором.
Осложнения
У страдающих болезнью Паллистера выявляются множественные пороки на фоне тяжелого неврологического дефицита, что обуславливает раннюю инвалидизацию таких пациентов. Осложнения синдрома связаны с задержкой речи и психического развития, из-за чего больным нужен постоянный уход, квалифицированная медицинская помощь. Неблагоприятный исход обычно наблюдается при тяжелых врожденных пороках, которые с трудом поддаются хирургической коррекции.
Диагностика
Своевременная постановка диагноза затруднена, что обусловлено большой редкостью синдрома Паллистера, наличием тканеспецифичного мозаицизма, что создает сложности при генетических анализах. Перед посещением генетика такие дети зачастую проходят осмотр у большого круга специалистов: педиатров, неврологов, кардиологов, эндокринологов. Для диагностики синдрома Паллистера назначаются следующие методы:
Лечение синдрома Паллистера
Консервативная терапия
Специальные методы лечения синдрома Паллистера пока не разработаны. Основу терапии составляют поддерживающие мероприятия, которые уменьшают клинические проявления болезни, стимулируют формирование отсутствующих психомоторных навыков, помогают адаптировать больного к жизни с инвалидностью. План терапии синдрома включает несколько направлений:
Оперативное лечение
Сердечные пороки, встречающиеся при синдроме, требуют оперативной коррекции, которая в основном проводится в первый год жизни ребенка. Сроки оказания кардиохирургической помощи определяются тяжестью нарушений кровообращения.
Прогноз и профилактика
С учетом отсутствия этиопатогенетических методов терапии синдрома Паллистера, тяжести клинической симптоматики и формирования инвалидности в раннем возрасте, прогноз неблагоприятный. Поскольку цель проводимых лечебных мероприятий — улучшение качества жизни пациентов, при их успешной реализации удается значительно развить психомоторные навыки. Специфическая профилактика синдрома отсутствует.
Нейрореабилитация детей: заболевания, методы, результаты
Врожденное или приобретенное поражение головного мозга часто звучит для родителей как приговор. Однако врачи утверждают: мозг обладает огромными резервными возможностями, нужно только правильно их использовать. У детей компенсаторные возможности особенно велики. Вернуть полноценное детство можно ребенку даже после тяжелой спинальной или черепно-мозговой травмы. Современные реабилитационные технологии позволяют улучшить качество жизни детей с ДЦП и хромосомными синдромами. В статье расскажем, как работает реабилитация при различных заболеваниях и какие зарубежные клиники предлагают новейшие восстановительные мероприятия.
Реабилитация детей с ДЦП. Современные методики позволяют людям с ДЦП доживать до преклонных лет, наслаждаясь всеми радостями жизни. Главное — как можно раньше начать терапию.
Курс формируется индивидуально в зависимости от проявлений болезни. Как правило, в него входят:
Грамотно подобранные занятия и поддержка родных помогают детям легче переносить заболевание и исключить развитие психологических расстройств. Когда ребенок вырастет, он сможет хорошо о себе позаботиться.
Реабилитация детей с синдромом Ретта. Девочки, которые им страдают, до года–полутора развиваются совершенно нормально. Поэтому диагноз производит на родителей эффект разорвавшейся бомбы. Невероятно сложно поверить в то, что твоему ребенку грозит тяжелая умственная отсталость.
Таким детям специалисты предлагают:
После реабилитационного курса психоэмоциональное состояние ребенка становится более стабильным. Насколько это возможно, улучшаются навыки общения, двигательные возможности и осанка. Тонус мышц снижается.
Реабилитация детей с синдромом Ангельмана. Подавляющее большинство людей даже не знают, что это за заболевание. Для них смеющийся без причины ребенок — просто счастливый маленький человек. Только родители и врачи знают, что скрывается за этим диагнозом. Помимо частого смеха синдром характеризуется гипервозбудимостью, отсутствием концентрации и внимания, серьезной задержкой развития. Дети растут, но психически и умственно остаются на уровне 2–3 лет.
Чтобы облегчить жизнь пациентов и особенно их близких, необходимо поддерживающее лечение, которое включает:
В результате ребенок становится более независимым и автономным.
В регулярных реабилитационных мероприятиях нуждаются также дети с синдромом Дауна, синдромом Мовата-Уилсона, синдромом Паллистер-Киллиана, синдромом Прадера-Вилли, синдромом Лежена, с миеломенингоцеле (спиной бифида), а также дети, страдающие от последствий нейроинфекций и травм.
Публикации в СМИ
Синдромы разные
Существует огромное количество различных синдромов, не вошедших в отдельные статьи или рассмотренных в рамках соответствующего заболевания. В этой статье даны краткие справки о ряде синдромов, в особенности выделенных в последнее время.
CADASIL (#125310, 19p13.2–p13.1, ген NOTCH3, Â ). Клинически: нарушения мозгового кровообращения, деменция, судороги, депрессия, нарушение двигательных функций, псевдобульбарный паралич, тетраплегия. КТ: инфаркты белого вещества мозга. Примечания • CADASIL — от англ. cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, артериопатия церебральная аутосомно-доминантная с подкорковыми инфарктами и лейкоэнцефалопатией • Известна форма CADASIL с алопецией и проявлениями дискоидной волчанки (600142, r ) • При мутациях и транслокациях генов NOTCH2 (600275, 1p13–p11) и NOTCH3 велик риск развития злокачественных новообразований. МКБ-10. I67.3 Прогрессирующая сосудистая лейкоэнцефалопатия.
CFC (от: cardiofaciocutaneous, кардио-кожно-лицевой, 115150, Â ) — множественные дефекты развития, проявляющиеся главным образом ВПС, деформациями лица и изменениями кожи (по типу ихтиозоподобного дерматоза, гиперкератоза и гиперпигментации). МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
GAPO (от: growth retardation — задержка роста, alopecia — алопеция, pseudoanodontia — псевдоанодонтия [задержка прорезывания зубов], progressive optic atrophy — прогрессирующая атрофия зрительных нервов, *230740, r ). Дополнительно: высокий выступающий лоб, гипоплазия средней трети лица, больших размеров открытый передний родничок, расширение вен головы, глаукома, кератоконус, стеноз хоан, задержка костного возраста, гипогонадизм, повторные инфекции. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
MASA (#303350, Xq28, ген L1CAM, 308840 [молекула адгезии клеток L1], À ). От: mental retardation — умственная отсталость, aphasia — афазия, shuffling gait — шаркающая походка, adducted finger приведённый большой палец кисти. Дополнительно: гиперрефлексия, отсутствие длинного и короткого разгибателя большого пальца кисти, укорочение туловища, выраженный поясничный лордоз. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
MIDAS (от: microphthalmos — микрофтальм, dermal aplasia — аплазия кожи, sclerocornea — помутнение роговицы, *309801, Xp22.31, гены MLS, MIDAS, À ). Дополнительно: линейные эритемы кожи головы и шеи, кисты глазницы, диафрагмальная грыжа, респираторный дистресс-синдром, аплазия прозрачной перегородки мозга. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
N (310465; ген NSX, À ) назван по первой букве фамилии больного. Клинически: умственная отсталость, нарушения зрения, поперечные складки верхнего века, глухота, аномалии ушей, крипторхизм, гипоспадия, лимфобластный лейкоз. Лабораторно: увеличение поломок хромосом, возможно, вследствие дефекта ДНК-полимеразы. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
TORCH — группа врождённых инфекций со сходными клиническими проявлениями, хотя симптомы могут варьировать по степени и времени проявления: токсоплазмоз (t), другие инфекции (o), краснуха (рубелла — r ), ЦМВ-инфекции (c) и герпес простой (h). МКБ-10. P37 Другие врожденные инфекционные и паразитарные болезни.
WAGR (комплекс WAGR, #194072, протяжённая делеция 11p13–11p16). От: Wilms tumor (опухоль Вильмса), Aniridia (аниридия), Genitourinary anomalies (аномалии мочевыделительного тракта, например псевдогермафродитизм, гонадобластома или дисплазия мочеполовая (#137357, 11p13, Â ) — атрезия уретры и мочеточников, двусторонний крипторхизм), mental Retardation (умственная отсталость). МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Авеллиса синдром — сочетание паралича мягкого нёба и голосовой мышцы на стороне патологического очага в ЦНС с центральным гемипарезом (гемиплегией) на противоположной стороне; относят к бульбарным альтернирующим синдромам; наблюдают при поражении продолговатого мозга на уровне двойного ядра. МКБ-10. G46.3* Синдром инсульта в стволе головного мозга (I60-I67+)
Акроренальный синдром (201310, r ). Клинически: клешня краба, клинодактилия, гипоплазия почек, почечная недостаточность. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Аладжила синдром — наследственное заболевание (#118450, 20p12, дефект гена AGS, Â ; протяжённая делеция 20p12.1–p11.23), проявляющееся множественными пороками развития на фоне гиперхолестеринемии и гиперлипидемии. У больных повышен риск развития артериальной гипертензии, рака печени и папиллярной карциномы щитовидной железы. Синонимы: холестаз и стеноз лёгочной артерии, дисплазия артерио-печёночная, синдром Аладжила–Уотсона. МКБ-10. Q44.7 Другие врождённые аномалии печени.
Апаллический синдром — состояние после выключения обширных областей коры при относительной сохранности ствола. Развивается быстро (при энцефалите, интоксикации, гипоксии, отёке, инсульте, ЧМТ, опухоли) или постепенно (на протяжении месяцев или лет в результате демиелинизирующих заболеваний, пресенильных атрофий мозга и др.). Клинически: нарушения двигательной сферы, речи, эмоций и памяти; мышечный тонус резко повышен; нарушение координации глазных яблок. Любые попытки фиксировать внимание больного безуспешны. На болевое раздражение возникает хаотическая двигательная активность. МКБ-10. G93.8 Другие уточнённые поражения головного мозга.
Апера синдром — комплекс наследственных аномалий (#101200, 10q26, мутация гена рецептора к фактору роста фибробластов FGFR2, Â ), включающий дизостоз черепа, гипертелоризм, экзофтальм, синдактилию, арковидное нёбо, отставание в умственном развитии и некоторые другие аномалии. Синонимы: акрокраниодисфалангия, акросфеносиндактилия, акроцефалосиндактилия. МКБ-10. Q87.0 Синдромы врождённых аномалий, влияющие преимущественно на внешний вид лица.
Ашера (Чарлза) синдром — группа синдромов слепоглухоты. Из 16 000 слепоглухих людей в США более половины, как полагают, имеют синдром Ашера, — комбинация прогрессирующей пигментной ретинопатии и врождённой нейросенсорной тугоухость. Идентифицировано несколько локусов на хр. 3, 10, 11, 14 и 21 (• тип 1A, USH1A, USH1, 276900, 14q32; • тип 1B, MYO7A, USH1B, DFNB2, DFNA11, 276903 (миозин VIIA), 11q13.5; • тип 1C, USH1C, 276904, 11p15.1; • тип 1D, USH1D, 601067, 10q; • тип 2, USH2A, 276901, 1q41; • тип 3, USH3, 276902, 3q21 q25). Клинически: пигментный ретинит с началом в возрасте 10 лет, потеря зрения, катаракта, глубокая врождённая нейросенсорная тугоухость, умственная отсталость, психозы, атаксия, нарушения речи. Примечание: внесиндромная глухота (#600060, 11q13, ген NSRD2, r ) аллельна с синдромом Ашера (тип IB): изолированная нейросенсорная тугоухость. МКБ-10. H91.3 Мутационная глухота, не классифицированная в других рубриках.
Ашера Барни синдром — наследуемая эритематозная пузырчатка (Сенира–Ашера синдром, 3q21-q25). МКБ-10. L10.4 Пузырчатка эритематозная.
Ашермана синдром — нарушение менструальной и детородной функций, обусловленное наличием внутриматочных синехий. МКБ-10. N85.6 Внутриматочные синехии.
Баньяна–Зонана синдром (#153480, 10q23.3, ген PTEN [MMAC1, 601728], Â ). Клинически: макроцефалия, скафоцефалия, множественные липомы, гемангиомы, мраморный оттенок кожи, телеангиэктазии, увеличение полового члена с выраженной пигментацией головки, большие длинна тела и масса плода при рождении, задержка моторного развития, нарушения координации, позднее развитие речи, умеренная умственная отсталость, грудь сапожника, повышенный риск развития внутричерепных опухолей, псевдоотёк диска зрительного нерва, гипертелоризм, высокое нёбо, миопатия, расширение больших пальцев кисти и стопы, аутоиммунный тиреоидит. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Барде–Бидла синдром (типы и гены: • 1, BBS1, 209901, 11q13; • 2, BBS2, 209900, 16q21; • 3, BBS3, 600151, 3p13 p12; • 4, BBS4, 600374, 15q22.3 q23). Клинически: ожирение, пигментная ретинопатия, полидактилия, синдактилия, брахидактилия, умственная отсталость, гипогенитализм, артериальная гипертензия, малые аномалии почек (расширение лоханок, кисты, дивертикулы чашечек, дольчатая почка), почечная недостаточность, СД, цирроз печени. Примечание. До недавнего времени в название синдрома Лоуренса–Муна включали имена Барде и Бидла. МКБ-10. Q87.8 Другие уточнённые синдромы врожденных аномалий, не классифицированные в других рубриках
Бекуитта–Видеманна синдром (типы и гены: • 130650, BWS, WBS, 11pter p15.4; • 130650, CDKN1C, KIP2, 600856, 11p15.5). Триада: пупочная грыжа, макроглоссия, гигантизм; часто при рождении отмечают гипогликемию, выраженную предрасположенность к развитию опухоли Вильмса. Лечение медикаментозное (ГК) и хирургическое (удаление опухолей). Диета. По возможности животные жиры следует заменять растительными, умеренное ограничение сливочного масла. Синоним: синдром EMG (от exomphalos, macroglossia, gigantism). МКБ-10. Q87.3 Синдромы врождённых аномалий, проявляющихся избыточным ростом [гигантизмом] на ранних этапах развития.
Биндера синдром (дисплазия максиллоназальная, *155050, Â или полигенное). Верхнечелюстно-носовая дисплазия, короткий нос, плоская переносица, короткий позвоночный столб, острый носогубной угол, выпуклая верхняя губа, аномалия прикуса, гипоплазия середины лица, отсутствие передней носовой ости, гипоплазия терминальных фаланг кисти, очаговые искривления позвонков, расщелины в позвонках. МКБ-10. Q87.0 Синдромы врожденных аномалий, влияющие преимущественно на внешний вид лица
Беспокойных ног синдром (акромелалгия наследуемая, *102300, Â ) — вазомоторный невроз; характерны покраснение, боль и отёчность пальцев конечностей, головные боли, рвота. МКБ-10. G25.8 Другие уточнённые экстрапирамидные и двигательные нарушения.
Бира–Стивенсона синдром (#123790, 10q25.3–10q26, мутация гена рецептора 2 фактора роста фибробластов FGFR2, Â ). Клинически: черепнолицевые аномалии (краниосиностоз, череп в форме трилистника), аномальные ушные раковины, складчатая кожа, чернеющий акантоз кожи, выросты кожи, аномалии гениталий и ануса; все описанные случаи спорадические. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Блума синдром — наследственное заболевание (210900, мутации гена ДНК–лигазы BLM, BS, 15q26.1) с пропорциональной пре- и постнатальной задержкой роста и умственного развития, повышенной чувствительностью к УФ-лучам, телеангиэктазиями, нарушениями пигментации кожи, склонностью к злокачественным новообразованиям, инфекциям, СД и нестабильностью хромосом. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Борьесона синдром (синдром Борьесона–Форсмана–Лемана, *301900, Xq26–q27, ген BFLS, À ). Клинически: тяжёлая умственная отсталость, судорожные припадки, гипогонадизм с небольшими яичками и постпубертатной гинекомастией, выраженное ожирение, узкие глазные щели, выступающие надбровные дуги, глубоко посаженные глаза, птоз, большие уши, узкие короткие пальцы кистей и стоп. МКБ-10. Q87.0 Синдромы врожденных аномалий, влияющие преимущественно на внешний вид лица
Брауэра синдром (*136500, Â ). Клинически: очаговая дисплазия кожи лица, складчатая или морщинистая кожа на висках, почти полное отсутствие подкожной клетчатки, атрофия кожи. МКБ-10. Q82.8 Другие уточнённые врожденные аномалии кожи
Бруннера синдром (*309850, недостаточность моноаминооксидазы А, КФ 1.4.3.4, Xp11.4–p11.23, ген MAO, À ). Клинически: изолированная умеренная умственная отсталость, агрессивное поведение. Лабораторно: недостаточность моноаминооксидазы А. МКБ-10. F70–F79 Умственная отсталость
Бука синдром (*112300, Â ). Гипергидроз, аплазия премоляров, гиподонтия, преждевременное поседение. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Бурхаве синдром — спонтанный разрыв нижнего отдела пищевода; разновидность синдрома Мэллори–Вейсса. МКБ-10. K22.6 Желудочно-пищеводный разрывно-геморрагический синдром
Ван дер Вуда синдром (*119300, 1q32, ген VWS, LPS, PIT, Â ). Расщелина губы и/или нёба и слизистые кисты нижней губы. МКБ-10. Q38.0 Врожденные аномалии губ, не классифицированные в других рубриках
Валленберга–Захарченко синдром — бульбарный синдром при поражении дорсолатеральной области продолговатого мозга в результате окклюзии задней нижней мозжечковой или позвоночной артерии; характерны гипестезия кожи лица, паралич мягкого нёба и голосовой мышцы, симптом Хорнера и атаксия — на стороне патологического очага, расстройство поверхностной чувствительности — на противоположной стороне « Синдром продолговатого мозга латеральный « Синдром ретрооливарный « Синдром тромбоза задней нижней мозжечковой артерии. МКБ-10. G46.3* Синдром инсульта в стволе головного мозга (I60-I67+)
Вольфа–Хиршхорна синдром — делеция короткого плеча хр. 4, сопровождающаяся множественными пороками развития (например, нос типа шлема греческого воина, антимонголоидный разрез глаз, ВПС и др.). Выражен дефект интеллекта. В 90% случаев делеция возникает de novo, остальные случаи обусловлены мозаицизмом или транслокациями у родителей; обнаружены случаи кольцевой хромосомы. Частота: 1 случай на 100 000 новорождённых. Диагноз основан на прямом определении кариотипа плода после проведения кордоцентеза или амниоцентеза. Синонимы: синдром делеции короткого плеча хр. 4, синдром 4р-. Примечание. Синдром впервые описан в 1965 г. двумя группами исследователей: немецкими генетиками во главе с Wolf, и американскими во главе с Hirschorn. МКБ-10. Q93.3 Делеция короткого плеча хр. 4.
Гоеминна синдром (синдром TKCR [от torticollis, keloids, cryptorchidism, renal dysplasy — кривошея, келоиды, крипторхизм, дисплазия почки]; *314300, Xq28, гены TKC, TKCR, À ). Дополнительно: пигментные родинки, олигоспермия, хронический пиелонефрит, односторонняя атрофия почки, варикозные вены, асимметрия лица. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Голденера с односторонними лучевыми аномалиями синдром (141400, 7p, ген GHS, Â ). Клинически: карликовость, кожные выросты в области угла нижней челюсти, укорочение нижней челюсти, различные дефекты верхней конечности (например, трёхфаланговый большой палец кисти, удвоение большого пальца кисти), расщелина губы, снижение слуха, микротия, атрезия наружного слухового прохода, множественные преаурикулярные выросты и ямки. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Гольденара синдром (164210, Â ) характеризуется эпибульбарным дермоидом, аномалией развития ушной раковины, микрогнатией, вертебральными и другими аномалиями « Окуло-аурикуло-вертебральная дисплазия. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Гольтца–Горлина синдром (*305600, Xp22.31, дефекты генов DHOF, FODH, À доминантное) — наследственная болезнь, проявляющаяся образованием резко отграниченных очагов истончённой гиперпигментированной кожи, дистрофией ногтей, гипотрихозом, аномалиями развития глаз, гортани, сердца и скелета. Синонимы: гипоплазия дермальная фокальная, аномалия наследственно-семейная эктомезодермальная. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Градениго синдром — сочетание симптомов гнойного среднего отита с парезом или параличом наружной прямой мышцы глаза, болями по ходу тройничного нерва; возникает при ограниченном менингите в области пирамиды височной кости. МКБ-10. H70.2 Петрозит.
Де Морсье синдром — комплекс сенсорных и моторных симптомов при травме, интоксикациях, врождённых дефектах или воспалении диэнцефальной области мозга, в т.ч. врождённая гипоплазия зрительного нерва вследствие аплазии ганглиозных нейронов сетчатки; может сочетаться с дефектами срединных структур промежуточного мозга (септо-оптическая дисплазия). МКБ-10. Q04 Другие врожденные аномалии [пороки развития] мозга.
Синдром дисморфии Симпсона (#312870, Xq26, мутации гена глипикана 3 [300037, GPC3, SDYS, SGB], À ). Клинически: внутриутробное и послеродовое ускоренное физическое развитие без нарушений интеллекта, непропорционально большая голова, грубые черты лица, выступающая челюсть, широкая спинка и вздёрнутый кончик носа, катаракта, гипертелоризм, монголоидный монголоидный (опущены внутренние углы глазных щелей) разрез глаз, отслойка сетчатки, специфическая форма ушных раковин, большой рот, центральная расщелина нижней губы, подслизистая расщелина нёба, высокое нёбо, короткая шея, «куриная грудь», дополнительные рёбра, добавочные соски, врождённая диафрагмальная грыжа, ДМЖП, стеноз лёгочной артерии с развитием лёгочной гипертензии, аритмии, дивертикул Меккеля, незавершённый поворот кишечника, спленомегалия, увеличенные диспластические почки, крипторхизм, гипоспадия, низкий тембр голоса, развёрнутые крылья подвздошной кости, сужение крестцово-подвзошных щелей, гипогликемия в период новорождённости, повышенный риск развития эмбриональных опухолей, высокая перинатальная и детская смертность. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Жакку синдром — полиартрит с внутримышечным фиброзом, контрактуры и анкилозы (болезнь развивается преимущественно у подростков). МКБ-10. M12.0 Хроническая постревматическая артропатия [Жакку].
Жёна синдром (*208500, 12p12.2–12p11.21, дефект гена ATD, r ) — наследственная патология, заканчивающаяся фатально в период новорождённости. Клинически: недоразвитая грудная клетка, гипоплазия лёгких, дыхательная недостаточность (синдром может проявляться респираторным дистресс-синдромом), полидактилия, дегенерация сетчатки, синдром мальабсорбции, хронический нефрит, кисты почки и поджелудочной железы, дефекты тазовых костей. Синонимы: дистрофия асфиктическая торакальная новорождённых, дистрофия торакально-тазово-фаланговая, дисплазия удушающая торакальная, хондродистрофия удушающая торакальная. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
Запертого человека синдром — отсутствие адекватной реакции на внешние (в т.ч. словесные) стимулы из-за тетраплегии и паралича бульбарной, мимической и жевательной мускулатуры (впервые описан ИН Филимоновым в 1923 г. и ЛВ Блуменау в 1924 г.). Быстро развивающиеся обширные поражения кортикоспинальных и кортиконуклеарных путей на уровне моста мозга при сохранности структур покрышки. Наблюдают при тромбозе базилярной артерии, остром стволовом энцефалите или остром полирадикулоневрите. Ясное сознание при полной утрате всех двигательных функций и речи, но функция отдельных глазодвигательных мышц иногда остаётся сохранной « Синдром отсутствия двигательных функций « Синдром изоляции. МКБ-10. I65.1 Закупорка и стеноз базилярной артерии
Запястный синдром (синдром канала запястья, наследуемая форма: *115430, Â ). Утолщение поперечной связки запястья, невропатия срединного нерва вследствие сдавления в суженном канале запястья, тендосиновит, недостаточность витамина B6, начало в раннем возрасте. МКБ-10. G56.0 Синдром запястного канала.
Ирвина–Гасса синдром — кистозный отёк жёлтого пятна, связанный с гиперпроницаемостью капилляров сетчатки. МКБ-10. H31 Другие болезни сосудистой оболочки глаза
Кальманна синдром обусловлен недостаточной секрецией гонадотропинов и проявляется гипогонадизмом в сочетании с аносмией из-за сопутствующей агенезии обонятельных долей мозга • Синдром Кальманна 1 (*308700, Xp22.3, дефект гена KAL1, À ). Клинически: гипогонадизм и аносмия. Лабораторно: дефицит гонадолиберина, нарушение секреции ФСГ и ЛГ, резистентность клеток Ляйдига к гонадотропинам • Синдром Кальманна 2 (*147950, Â ). Дополнительно: низкий рост, умственная отсталость, ВПС, нейросенсорная тугоухость • Синдром Кальманна 3 (*244200, r ). Дополнительно: расщелина губы, расщелина нёба, гипотелоризм • Синдром Кальманна со спастической параплегией (308750, Xp22.3, дефект гена KAL1, À ). МКБ-10 • E23.0 Гипопитуитаризм
Карша–Нойгебауэра синдром (183800). Клинически: катаракта, ретинопатия, нистагм, эктродактилия. МКБ-10 Q15.8 Другие уточнённые пороки развития глаза. Примечание. Эктродактилия — аномалия развития: отсутствие одного или нескольких пальцев или кисти. К эктродактилии относят также деформации типа клешня краба (бидактилия — наличие только I и V пальцев или только двух пальцев на руке, реже стопе « двупалость). В каталоге OMIM эти аномалии развития зарегистрированы как отдельные нозологические единицы (например, синдром EEC: 129900, 7q11.2 q21.3) с разными типами наследования и как составляющая других заболеваний.
Каста синдром — хондроматоз пястных и/или плюсневых костей и фаланг пальцев, сочетающийся с венозными гемангиомами и флеболитами. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Кауфмана синдром (*244450, r ). Клинически: внутриутробная и последующая задержка роста и развития, умственная отсталость, микроцефалия, гипертелоризм, эпикантус, птоз, блефарофимоз, нистагм, косоглазие, амблиопия, миопия, микрогнатия, высокое нёбо, низко посаженные ушные раковины, лордоз, суставные контрактуры, вялая кожа. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем. Примечание. Блефарофимоз — укорочение глазной щели, обусловлено срастанием краёв век у наружного угла глаза при хроническом конъюнктивите, часто признак наследственной патологии.
Кисте-стопо-генитальный синдром (#140000, 7p15–p14.2, ген HOXA13, 142959, Â ). Клинически: удвоение полового тракта (двурогая матка, продольная влагалищная перегородка) у женщин, недержание мочи, частые инфекции мочевых путей, пузырно-мочеточниковый рефлюкс, гипоспадия с искривлением полового члена, маленькие стопы, короткий большой палец кистей и стоп, клинодактилия, гипоплазия мышц возвышения большого пальца, косоглазие, укорочение первых костей пястья и плюсны, короткие пятые пальцы с клинодактилией, слияние трапециевидной и ладьевидной костей запястья, слияние клиновидной и ладьевидной костей стоп. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем
Китайского ресторана синдром. Появление боли в груди, гиперемия лица, чувство жжения в различных участках тела после приёма в пищу L-глутамата натрия. МКБ-10. E72.8 Другие уточнённые нарушения обмена аминокислот.
Клиппеля–Треноне–Уэбера синдром (невус варикозный остеогипертрофический, 149000, Â ; возможен мозаицизм) — аномалия развития глубоких магистральных вен конечностей, проявляющаяся гипертрофией конечностей, варикозным расширением поверхностных вен, гемангиомами, пигментными пятнами. Клинически: огромные гемангиомы кожи, асимметричная гипертрофия конечностей, аневризмы почечных артерий, гемангиомы почек, таза, матки, водянка плода. МКБ-10. Q87.2 Синдромы врождённых аномалий, вовлекающих преимущественно конечности.
Конского хвоста синдром. Атоничный мочевой пузырь, нарушение функции дефекации, периферический нижний парапарез. МКБ-10. G83.4 Синдром конского хвоста.
Кошачьего глаза синдром (#115470, ген CECR, CES, 22q11). Уменьшение размеров уха, колобома радужки и вертикальный зрачок (подобный зрачку кошки), микроофтальмия, ВПС, атрезия ануса, меккелев дивертикул, атрезия жёлчных путей, мальформации почек, возможна умственная отсталость. Синонимы: частичная тетрасомия хр. 22, синдром Шмида–Фраккаро. МКБ-10. Q99.8 Другие уточнённые хромосомные аномалии.
Кошачьего крика синдром обусловлен делецией хр. 5, в большинстве случаев возникающей de novo; сопровождается множественными пороками развития. Характерна аномалия гортани, обусловливающая необычно высокий тембр крика в период новорождённости. Многие больные доживают до взрослого возраста. Частота: 1 случай на 50 000 новорождённых. Синонимы: синдром делеции короткого плеча хр. 5, синдром cri du chat. МКБ-10. Q93.4 Делеция короткого плеча хр. 5.
Краниофациальный с глухотой и дефектами рук синдром (#122880, 2q35, ген гомеозисный PAX3 [193500], Â ). Клинически: плоский лицевой профиль, гипертелоризм, гипопластический нос с щелевидными ноздрями, гипоплазия верхней челюсти. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
Крузона синдром (• #123500, 10q25.1–q25.2, дефект гена каспазы CASP7; Â • с чернеющим акантозом, дефект гена рецептора фактора роста фибробластов FGFR3, 134934, 4p16.3). Клинически: краниосиностоз, выступающие глаза, гипертелоризм, косоглазие, вздёрнутый кончик носа, короткая верхняя губа, гипоплазия верхней челюсти, прогнатия, выраженные пальцевые вдавления на своде черепа. МКБ-10. Q75.1 Краниофациальный дизостоз.
Крювелье–Баумгартена синдром. Цирроз печени, незаращение пупочной вены или околопупочных вен, варикозные расширения вен области пупка (голова медузы). МКБ-10. K74.6 Другой и неуточнённый цирроз печени.
Коффин–Лаури синдром (#303600, Xp22.2, ген RPS6KA3, À доминантное). Клинически: умственная отсталость, гипотония, гидроцефалия, шейная радикуломиелопатия, широкий нос с вывернутыми ноздрями, нейросенсорная тугоухость, большие уши, гипертелоризм, антимонголоидный разрез глазных щелей, широкие брови, большой рот, гипоплазия верхней челюсти, суженные легкорастяжимые пальцы, широкие предплечья, плоскостопие, шейный лордоз, кифосколиоз, низкий рост, гидронефроз, кишечный дивертикулёз, недостаточность митрального клапана, панацинозная эмфизема, преждевременное выпадение молочных зубов, задержка костного возраста, кальциноз связок, сужение расстояния между позвонками. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
Коэна синдром (*216550, 8q22–q23, ген COH1, r ). Клинически: ожирение, мышечная гипотония, умеренная умственная отсталость, микроцефалия, судорожные приступы, высокая переносица, гипоплазия коренных зубов, короткий губной желобок, антимонголоидный разрез глаз, хориоретинальная дистрофия, снижение остроты и полей зрения, пигментные отложения на сетчатке, атрофия зрительных нервов, изоэлектрическая электроретинограмма, отслойка сетчатки, выступающие центральные резцы верхней челюсти, открытый рот, крупные губы, пролапс митрального клапана, узкие кисти и стопы с удлинёнными пальцами, желудочно-пищеводный рефлюкс, диафрагмальная грыжа. Синонимы: синдром Пеппера, гипотония с ожирением и выступающими резцами. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
Ларсена синдром (150250, 3p21.1–p14.1, гены LRS1, LAR1, Â ; для рецессивной формы [245600] дополнительно характерна низкорослость). Клинически: множественные врождённые вывихи, выступающий лоб, плоское лицо, гипертелоризм, сколиоз, брахидактилия, полая стопа, цилиндрические пальцы, добавочные запястные кости, короткие терминальные фаланги, глухота, добавочные точки окостенения, патологические позвонки.
Леша–Найена синдром (*308000, КФ 2.4.2.8, Xq26–q27.2, дефект гена HPRT, À рецессивное) проявляется только у мальчиков повышенной экскрецией мочевой кислоты и уратов, хореоатетозом, умственной отсталостью, спастическими центральными парезами, приступами агрессивного поведения со склонностью к членовредительству вследствие абсолютной недостаточности гипоксантин-гуанин фосфорибозил трансферазы. При частичной недостаточности фермента — острый подагрический артрит, нефролитиаз. МКБ-10. E79.1 Синдром Леша–Найена.
Линча синдром. Из множества семейных форм карцином группа Линча выделила два типа наследуемого ( Â ) неполипозного рака толстой и прямой кишки. Не менее 5% колоректальных карцином (особенно с ранним началом) приходится на эти два синдрома. Соответствующие гены расположены в хр. 2 (MSH2, 2p16–p15) и 18 (CRCR1; CRC18; DCC, 18q11–q12) • Тип I: рак толстой кишки, преимущественно правосторонний • Тип II: карцинома толстой кишки, а также рак эндометрия, яичника и поджелудочной железы (в сочетании с колоректальным раком или самостоятельно). Обнаружена корреляция с Аг группы крови Кидд (локус Jk), а также сходная локализация локусов DCC и Jk. МКБ-10. C18 Злокачественное новообразование ободочной кишки.
«Ломкой кожи» синдром (*601975, 1q32–q44, ген плакофилина-1 PKP1, r ) — мутация в гене белка плакофилина-1, играющего важную роль в формировании цитоскелета клеток и межклеточных взаимодействиях. Десмосомы между клетками эпидермиса плохо сформированы, межклеточные пространства расширены. МКБ-10. Q82 Другие врожденные аномалии [пороки развития] кожи.
Лоуренса–Муна синдром (*245800, r ) — наследственный пигментный ретинит с неврологической и эндокринной симптоматикой. Клинически: пигментная ретинопатия, гипогенитализм, гипогонадизм, умственная отсталость, спастическая параплегия. Дифференциальная диагностика: синдром Барде–Бидла. МКБ-10. Q87.8 Другие уточнённые синдромы врождённых аномалий, не классифицированные в других рубриках.
Лоу синдром — наследуемое нарушение обмена инозитол 1,4,5-трифосфата (*309000, Xq26-1, дефект гена OCRL, À ), характеризующееся поражением глаз (катаракта и глаукома), отставанием в умственном развитии, повышением выделения с мочой органических кислот, почечной недостаточностью и витамин D-резистентным рахитом. Синоним: синдром окуло-церебро-ренальный. МКБ-10. E72.0 Нарушения транспорта аминокислот.
Малуфа синдром (212112, r ; 115200, Â ; 302045, À ) — застойная дилатационная кардиомиопатия, овариальный дисгенез, гипергонадотропный гипогонадизм « Дилатационная кардиомиопатия с гипергонадотропным гипогонадизмом.
Маринеско–Шёгрена синдром (*248800, синдром MSS, ген MSS, r ). Клинически: мозжечковая атаксия, умственная отсталость, дизартрия, спастичность, врождённая катаракта, нистагм, косоглазие, микроцефалия, малый рост, гипергонадотропный гипогонадизм, мышечная слабость, кифоз, сколиоз. Лабораторно: атрофия коры мозжечка.
Мёбиуса синдром — наследственный паралич нервов (преимущественно черепных) (*157900, 13q12.2–q13, Â ; • тип 2, MBS2, 601471, 3q21 q22). Клинически: затруднённые сосание, глотание и сокращения мимических мышц, одно- или двусторонний птоз, слабость жевательной мускулатуры, атрофия языка, периферические невропатии; возможны тугоухость, отставание в умственном развитии, гипогонадотропный гипогонадизм, гипоплазия нижней челюсти, артрогрипоз и другие дефекты. МКБ-10. Q87.0 Синдромы врождённых аномалий, влияющие преимущественно на внешний вид лица.
Мейга синдром — формирование асцита и гидроторакса при фиброме яичника (реже при опухолях малого таза). Синонимы: синдром асцита-плеврального выпота овариального генеза, синдром Демона–Мейга, синдром Мейга–Косса. МКБ-10 • C56 Злокачественное новообразование яичника
Меккеля синдром — наследуемый комплекс дефектов развития (*249000, 17q21–q24, дефект гена MKS, r ) с высокой летальностью в период новорождённости, характеризующийся деформациями костей черепа, полидактилией и синдактилией, катарактой, поликистозом почек, дисгенезией гонад, гипоплазией мочеточников и мочевого пузыря, пороками развития печени, селезёнки, надпочечников и лёгких. Частота: не менее 1:100 000, значительно выше среди татар и финнов — 1:9 000. Синонимы: синдром Грубера, синдром Меккеля–Грубера, дизэнцефалия спланхнокистозная. МКБ-10. Q61.9 Синдром Меккеля–Грубера.
Мелькерссона–Розенталя синдром (*155900, 9p11, ген MROS, Â ). Клинически: сочетание неврита лицевого нерва, отёка и уплотнения лица и губ со складчатым языком, реже с парестезиями пальцев и эпизодами расстройства глотания, начало в детстве или юности; течение рецидивирующее. МКБ-10. G51.2 Синдром Россолимо–Мелькерссона–Розенталя.
Менкеса синдром — врождённый фатальный дефект метаболизма меди (*309400, Xq12–q13, дефекты генов, кодирующих катион-транспортирующую АТФазу ATP7A, MNK, MK, OHS, 300011, Xq12 q13, À рецессивное), характеризующийся повышенным содержанием меди в тканях (кроме печени). Клинически: слабо пигментированные, редкие курчавые волосы, судороги, физическое и отставание в умственном развитии, прогрессирующее поражение мозга, гипоплазия гонад. Синонимы: болезнь курчавых волос, болезнь Менкеса, трихополиодистрофия. МКБ-10. E83.0 Нарушения обмена меди.
Мийара–Гублера синдром — разновидность альтернирующей гемиплегии вследствие инфаркта моста с поражением VI и VII черепных нервов и волокон кортикоспинального тракта. Синоним: паралич Гублера. МКБ-10. G81.9 Гемиплегия неуточнённая.
Микулича синдром — сочетанное увеличение слёзных и всех слюнных желёз с понижением их секреторной функции. МКБ-10. K11.8 Другие болезни слюнных желёз.
Мишера синдром — наследственная болезнь, характеризующаяся сочетанием акантоза кожи чернеющего, СД, слабоумия, инфантилизма, складчатой пахидермии, гипертрихоза и деформации зубов.
Морщинистой кожи синдром (*278250, 2q32, ген WSS, r ). Клинически: врождённая морщинистость кожи кистей и стоп, слабо развитая скелетная мускулатура, гипотония мышц, крыловидные лопатки, подчёркнутая венозная сеть на груди, умственная отсталость, микроцефалия. Лабораторно: патологические эластичные волокна при биопсии кожи.
Мэллори–Вейсса синдром — разрыв слизистой оболочки желудка или нижнего конца пищевода, сопровождающийся кровотечением или пенетрацией в средостение, с сопутствующим медиастинитом; обычно происходит при сильных позывах к рвоте или во время рвоты. У большинства больных кровотечение останавливается самостоятельно. Этиология • Рвота на фоне алкогольной интоксикации • Диафрагмальная грыжа • Атрофический гастрит • Эзофагит. Лечение • Вазопрессин • При длительно продолжающемся кровотечении показано оперативное лечение: лапаротомия, высокая гастротомия и прошивание кровоточащих сосудов. МКБ-10–10. K22.6 Желудочно-кишечный разрывно-геморрагический синдром.
Мюир–Торр синдром (#158320, 2p22–p21, гены MSH2, COCA1, FCC1, 120435, Â ). Клинически: сальные опухоли кожи, карцинома двенадцатиперстной и толстой кишки, гортани, преждевременная менопауза.
Негели синдром (*161000, синдром Негели–Франческетти–Ядассона, Â ). Клинически: ретикулярная пигментация кожи, гипогидроз, ладонно-подошвенный гиперкератоз, гипоплазия дерматоглифических узоров ладоней, жёлтая эмаль зубов, неправильная форма зубов, добавочные зубы, ониходистрофия, врождённая отслойка ногтя большого пальца стопы, редкие волосы; пигментация кожи исчезает при наступлении половой зрелости.
Недержания пигмента синдром (1 тип, 308300, Xp11.21; 2 тип, *308310, Xq28, À доминантное) — наследственный дерматоз, характеризующийся появлением на боковых поверхностях тела вскоре после рождения пигментных пятен причудливой формы (как правило, исчезают к 20 годам жизни); сочетается с аномалиями развития зубов, волос, глаз и т.д. Синонимы: синдром Блоха–Сульцбергера, Сименса–Блоха пигментный дерматоз, Блоха–Сульцбергера меланобластоз, дерматоз пигментный, меланоз дермы дегенеративный, невус семейный хроматоформный. МКБ-10. Q82.3 Недержание пигмента [incontinentia pigmenti].
Неподвижных ресничек синдром — дефекты белков, входящих в состав мерцательных ресничек и их аналогов (например, эпителий дыхательных путей, сперматозоиды), приводят к невозможности функционирования тубулин-динеинового хемомеханического преобразователя и неподвижности ресничек. Клинически: хроническая патология воздухоносных путей и респираторного отдела лёгкого (риниты, синуситы, носовые полипы, трахеобронхиты, бронхоэктазии, пневмонии), а также неподвижность сперматозоидов. Известно несколько наследственных форм синдрома (все r ) • Дефекты динеина микротрубочек (*242650, 6р, ген ICS1) • Транспозиция микротрубочек (*215520, r ) • Дефекты динеиновых ручек (*242670) • Избыточно длинные реснички (242680). МКБ-10. Q87 Другие уточнённые синдромы врождённых аномалий, затрагивающих несколько систем.
Нунан синдром (*163950, 12q22–qter, ген NS1, Â ) — так называемый мужской вариант синдрома Тёрнера (мужской фенотип при синдроме Тёрнера). Клинически: ВПС (в особенности стеноз лёгочного ствола), дисморфия шеи, грудной клетки и век. МКБ-10. Q87.1 Синдромы врождённых аномалий, проявляющихся преимущественно карликовостью.
Нэнси–Суини–Инсли синдром (215150, мутация гена коллагена COL11A2, 6p21.3, r ). Седловидный нос, прогрессирующая тугоухость, расщепление нёба, платиспондилия, слияние костей запястья, увеличение размеров эпифизов. Синонимы: хондродистрофия с нейросенсорной тугоухостью, синдром Нэнси–Инсли, хондродисплазия Нэнси–Суини, ото-спондило-мегаэпифизарная дисплазия.
Опитца синдром — комплексы наследственных врождённых пороков развития (тип 1, *300000, Xp22, гены OGS1, BBBG1, GBBB1, OSX, À ; тип 2, *145410, 22q11.2, гены OGS2, BBBG2, GBBB2, Â ). Клинически: телекант (смещение внутренних углов глазных щелей латерально при нормально расположенных глазницах.), гипертелоризм, расщелина губы и/или нёба, дисфагия, желудочно-пищеводный рефлюкс, нарушение моторики желудка, неперфорированный задний проход, гипоспадия, крипторхизм, расщеплённая мошонка, аномалии почек и мочеточников, хриплый крик, ларинготрахеопищеводные свищи, умственная отсталость, агенезия мозолистого тела, ВПС, пупочная грыжа, широкая полость прозрачной перегородки мозга на МРТ. Синонимы: синдром гипоспадии–дисфагии, телекант с сопутствующими аномалиями, синдром Опитца–Фриаса, синдром G, синдром BBB. МКБ-10. Q87.1 Синдромы врождённых аномалий, проявляющихся преимущественно карликовостью.
Осмотической демиелинизации синдром — ограниченное разрушение миелина в области основания моста мозга; может быть связан с голоданием, алкоголизмом, гипотонической дегидратацией; клинически проявляется тетраплегией, псевдобульбарным параличом, иногда судорогами. « Центральный мостовой миелинолиз. МКБ-10. G37.2 Центральный понтинный миелинолиз.
Отмены ГК синдром — гипофизарно-адренокортикальная недостаточность, развивающаяся после длительного применения больших терапевтических доз ГК; эмоциональные расстройства, особенно после стресса, на протяжении более чем года после прекращения глюкокортикоидной терапии.
Парри–Ромберга синдром (141300, Â ) — форма локализованной склеродермы. Клинически: медленно прогрессирующая атрофия мягких тканей одной половины лица, участки вдавления костей лица, контралатеральная джексоновская эпилепсия, невралгия тройничного нерва, мигренеподобные головные боли, гиперпигментация кожных покровов или витилиго, алопеция, гемиатрофия языка, аномалии прикуса, энофтальм, патология рефракции, гетерохромия радужек.
Партингтона синдром (*301220, Xp22–p21, ген PDR, À ) — отложения амилоида в сосочковом слое кожи, очаги гиперпигментации, задержка развития, множественные расстройства функций ЦНС и внутренних органов. МКБ-10. L99.0* Амилоидоз кожи (E85.-+)
Паттерсона–Стивенсона–Фонтэна синдром (183700, Â ). Клинически: гипоплазия нижней челюсти, ретрогнатия, расщелина нёба, дефекты пальцев.
Пиквикский синдром (синдром ожирения–гиповентиляции) — необычное уродующее ожирение, проявляющееся гиповентиляцией (с артериальной гипоксемией и гиперкапнией), сонливостью и некоторой дебильностью. Примечание. Персонаж («жирный парень») из книги «Записки Пиквикского клуба» Ч. Диккенса. МКБ-10. E66.2 Крайняя степень ожирения, сопровождаемая альвеолярной гиповентиляцией. OMIM 257500 ( r ).
Персистенции мюллеровых протоков синдром — мужской псевдогермафродитизм, вызванный нарушением подавления развития мюллеровых протоков у мальчиков (тип I, #261550, 19p13.3–p13.2, мутации гена мюллеровского ингибирующего фактора MIF, 600957, r ; мутации гена рецептора антимюллеровского фактора [тип 2, #261550, 12q13, ген AMHR, 600956, r ]). Клинически: двусторонний крипторхизм, паховая грыжа, содержащая матку и маточные трубы.
Пламмера–Винсона синдром (Патерсона–Келли синдром, синдром сидеропенический) — заболевание, предположительно обусловленное дефицитом рибофлавина и фолиевой кислоты. Клинически: дисфагия, атрофия слизистой оболочки ЖКТ, дистрофия ногтей, гипохромная анемия, уменьшение содержания железа в крови. МКБ-10. D50.1 Сидеропеническая дисфагия.
Плацентарной недостаточности синдром — гипотрофия и гипоксия плода вследствие дегенеративных изменений в плаценте (как правило, при переношенной беременности). МКБ-10. O43 Плацентарные нарушения.
Приводящей петли синдром. Нарушение пассажа содержимого приводящей кишечной петли, проявляющееся болями в правом подреберье и рвотой жёлчью; позднее осложнение резекции желудка.
Протея синдром (176920, Â ) проявляется множественными аномалиями развития (например, частичный гигантизм кистей и стоп, синдактилия, лимфангиомы, липомы, эпидермальные невусы, гипертрофия кожи подошв, депигментация/гиперпигментация, подчёркнутая венозная сеть на грудной клетке, опухоли склеры, пороки черепа и внутренних органов). Дифференциальная диагностика • Синдром Клиппеля–Треноне–Уэбера • Маффучи синдром. Примечание. Греческий бог Протей мог по желанию изменять свою форму.
Пустого турецкого седла первичный синдром (130720, Â ). Клинически: дефект спинки турецкого седла, увеличение размеров турецкого седла, внутреннего слухового прохода и отверстия зрительного нерва, добавочные косточки в лямбдовидном шве, остеосклероз, приподнятые углы нижней челюсти, аномалии развития спинного мозга, мозжечка и коры, множественные спинномозговые грыжи, умеренная задержка роста, гипоплазия лица, антимонголоидный разрез глаз, гипоплазия дёсен, высокое арковидное нёбо, гипоплазия нижней челюсти, увеличенное большое затылочное отверстие, платибазия, базилярная импрессия, расширение спинномозгового канала и внутрипозвонковых отверстий, аномалии тел позвонков.
Пьера Робена синдром (261800, Â и À ) может встречаться изолированно и в составе некоторых синдромов, включая трисомию 18. Клинически: глоссоптоз, микрогнатия, расщелина нёба, врождённый респираторный дистресс-синдром. МКБ-10. Q87.0 Синдромы врождённых аномалий, влияющие преимущественно на внешний вид лица.
Раппа–Ходжкена синдром (*129400, Â ). Клинически: ангидротическая эктодермальная дисплазия, расщелина губы и нёба, маленький рот, расщепление нёбного язычка, узкий нос, грубые, сухие и жёсткие волосы, раннее облысение, гиподонтия, птоз, аномалии слёзного протока, сужение слухового прохода, дисплазия евстахиевых труб, дистрофия ногтей, гипоспадия. Синонимы: эктодермальная дисплазия ангидротическая с расщелиной губы и нёба.
Распада опухоли синдром — гиперфосфатемия, гипокальциемия, гиперкалиемия и гиперурикемия, возникающие при химиотерапии злокачественной опухоли; следствие высвобождения внутриклеточных продуктов при лизисе клеток.
Резистентных яичников синдром наблюдают у женщин с кариотипом 46,ХХ при наличии аменореи и дефекта мембранных рецепторов яичников. Клинически: аменорея в сочетании с гипергонадотропизмом и нормальными овариальными фолликулами. Содержание гонадотропинов повышено, поскольку яичники нечувствительны к действию гонадотропинов, не секретируют гормонов и не угнетают функцию гипофиза. Вследствие возможной малигнизации, ХY-гонаду необходимо удалить до полового созревания или сразу после диагностики. Синоним: Savage (Саваж) синдром (по фамилии больной). МКБ-10. E28.8 Другие виды дисфункции яичников.
Робиноу синдром — наследственная остеохондродисплазия с врождённой карликовостью, различают две формы: • доминантная (*180700, Â ) — более частая • рецессивная (*268310, r ) — более тяжёлая. Клинически: остеохондродисплазия, мезомелическая дисплазия, умеренная задержка роста, макроцефалия, выпуклый лоб, «лицо плода», микрогнатия, вдавленная переносица, гипертелоризм, гипопластичные гениталии, крипторхизм, полупозвонки, вдавленная грудина, брахидактилия, короткие предплечья, широкий большой палец кисти, аномалия запястья типа Маделунга, неровные зубы. Лабораторно: повышение уровня ФСГ. Синонимы: карликовость Робиноу, синдром «лица плода». МКБ-10. Q87.1 Синдромы врожденных аномалий, проявляющихся преимущественно карликовостью.
Росселли–Гульенетти синдром (*225000, r ). Клинически: ангидроз, гипотрихоз, микродонтия, дисплазия ногтей, подколенный и перинеальный птеригиум (крыловидные складки кожи), расщелина губы/нёба, аномалии ушных раковин, деформированные пальцы кистей и стоп, синдактилия, аномалии почек, умственная отсталость, добавочные соски. Синонимы: эктодермальная дисплазия, расщелина губы и нёба, синдром Злотогора–Огура.
Ротмунда–Томсона синдром (*268400, хр. 8, ген RTS, r ). Клинически: пойкилодермия лица и конечностей, двусторонняя катаракта, дистрофия волос (ногтей и зубов), гипогонадизм, нарушения эндохондрального окостенения, артериосклероз и карликовость. Дополнительно: гиперпигментация кожи, телеангиэктазии, атрофический дерматоз, анемия, повышен риск остеогенной саркомы. Синонимы: атрофическая пойкилодермия и катаракта, дистрофия Ротмунда.
Рото-лице-пальцевой синдром — группа наследственных заболеваний, проявляющихся множественными врождёнными пороками развития (как правило, лица и пальцев). Различают несколько типов синдрома, например тип I (*311200, Xp22.3–p22.2, дефект гена OFD1, À доминантное). Клинически: умственная отсталость, расщелина верхней челюсти, широкий корень носа, маленькие ноздри, гипоплазия хрящей носа, срединная расщелина верхней губы, добавочная гиперпластичная уздечка губы, «дольчатый» язык с гамартомами, асимметричная расщелина нёба, синдактилия, брахидактилия, полидактилия, дизартрия, неуклюжая походка, гнёздная алопеция, поликистоз почек, почечная недостаточность, агенезия мозолистого тела на КТ, неравномерная минерализация костей кистей и стоп. МКБ-10. Q87 Другие уточнённые синдромы врождённых аномалий [пороков развития], затрагивающих несколько систем.
Рубинштейна синдром [#180849, 16p13.3, ген CREBBP (600140, коактиватор фактора транскрипции CREB), Â ] — широкие большие пальцы кистей и стоп, характерное лицо (косой разрез глазных щелей, клювовидный нос, короткая верхняя губа, выступающая нижняя губа) и олигофрения в сочетании с множественными пороками развития. Синонимы: Рубинштейна–Тейби синдром, синдром широкого большого пальца стоп и кистей. МКБ-10. Q87.5 Другие синдромы врождённых аномалий с другими изменениями скелета.
Саммерскилла синдром (*243300, недостаточность АТФазы типа Р, 18q21, r ) — внутрипечёночный доброкачественный холестаз. Клинически: зуд кожи, эпизоды желтухи, гепатомегалия, билиарный цирроз.
Свита синдром — множественные бляшкоподобные поражения кожи (лица, шеи, верхних конечностей), конъюнктивит, афтоподобные поражения слизистых оболочек, лихорадка, артралгии, сдвиг влево; полиморфноядерные лейкоциты в виде инфильтратов в коже; быстрая ремиссия под влиянием ГК. Преобладающий пол — женский. Синоним: Свита болезнь. МКБ-10. L98.2 Лихорадочный нейтрофильный дерматоз Свита
Сдавления верхней полой вены синдром чаще развивается при злокачественных опухолях средостения. Клинически: отёчность лица и верхней половины туловища, покраснение лица, набухание поверхностных вен, кашель, нехватка воздуха и повышение ВЧД.
Сениора–Локена синдром (*266900, r ) проявляется почечной дисплазией, прогрессирующей почечной недостаточностью, нефрогенным несахарным диабетом, пигментным ретинитом и дистрофией/аплазией сетчатки, мозжечковой атаксией, нейросенсорной тугоухостью и врождённым фиброзом печени. Синоним: дисплазия почек и аплазия сетчатки.
Смита–Лемли–Опитца синдром (*270400 [тип 1], #268670 [тип 2, аллельная мутация], 7q32.1, дефекты генов SLOS, SLO, D7SR, r ) — комплекс множественных врождённых аномалий, проявляющихся задержкой физического и умственного развития, дефектами черепа, мочевыделительной системы, ССС, лёгких и костной системы, а также катарактой, гипоспадией, крипторхизмом, что приводит к ранней младенческой смерти. Лабораторно: нарушения обмена холестерина. МКБ-10. Q87.1 Синдромы врожденных аномалий, проявляющихся преимущественно карликовостью.
Смита–Мейджениса синдром (#182290, 17p11.2, ген SMCR, протяжённая делеция). Клинически: брахицефалия, гипоплазия средней трети лица, прогнатия, хриплый голос, задержка психомоторного и физического развития, тенденция к самоповреждениям, расстройства сна, периферическая невропатия, снижение глубоких сухожильных рефлексов, плоскостопие, снижение болевого порога, глухота, расщелина твёрдого нёба, ВПС, дистрофия мышц ног, сколиоз, полупозвонки, половые аномалии.
Стиклера синдром — прогрессирующая близорукость в сочетании с дефектами развития позвонков и длинных трубчатых костей, вызванная мутациями в трёх различных локусах (• тип I, 108300, COL2A1 ( a 1 цепь коллагена II, 120140, 12q13.11 q13.2); • тип II, 184840, COL11A2 ( a 2 цепь коллагена типа XI, 120290, 6p21.3); • тип III, COL11A1, 120280, 1p21). Другие проявления: глухота, пролапс митрального клапана. Синоним: артроофтальмопатия наследуемая прогрессирующая.
Таунса–Брокса синдром (#107480, 16q12.1, ген SALL1 [HSAL1, TBS, фактор транскрипции, 602218], Â ). Клинически: нейросенсорная тугоухость, преаурикулярные ямки, аномальные ушные раковины типа «сатира», стеноз или незаращение заднего прохода, подчёркнутый шов промежности, ректовагинальные и ректопромежностные свищи, дефекты кисти и стопы, дисплазия лучевой кости, гипоплазия почек, ДМЖП.
Торакоабдоминальный синдром (*313850, пентада Кэнтрелла, Xq25–q26.1, ген TAS, À ) — комплекс врождённых пороков развития дизрафического ряда. Клинически: расщелина нёба и губы, кистозная гигрома, диафрагмальная грыжа, пороки формирования грудины, ВПС, транспозиция магистральных сосудов, открытый артериальный проток, гипоплазия лёгких, грыжи передней брюшной стенки, эмбриональная грыжа пупочного канатика, гидроцефалия, анэнцефалия, агенезия почек, гипоспадия. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
Труссо синдром — ДВС у онкологических больных, часто наблюдают при аденокарциноме поджелудочной железы, желудка и предстательной железы, реже — при других аденокарциномах.
Тюрко синдром (• 276300, APC, GS, FPC [рак толстой кишки], 114500, 5q21 q22; • с глиобластомой; • с глиобластомой 276300, MLH1, COCA2 [рак толстой кишки семейный неполипозный], 120436, 3p21.3; • 276300, PMS2, PMSL2, 600259, 7p22). Клинически: полипоз (аденоматоз) ободочной и толстой кишок, аденокарцинома толстой кишки и желудка, центральная узелковая гиперплазия печени, глиома, глиобластома, астроцитома, пятна цвета «кофе с молоком», множественные липомы.
Углевододефицитных гликопротеинов синдром (CDG1, 212065, 16p13.3 p13.2, тип I [601785, ген PMM2]) развивается при дефектах посттрансляционной модификации в зоне Гольджи. Клинически: задержка психомоторного развития, общая мышечная гипотония, гипорефлексия, миксома предсердия, лёгкое ожирение, косоглазие, гипоплазия мозжечка, гиперпигментация кожи. Синонимы: болезнь Кушинга с миксомой предсердия и гиперпигментацией, миксома-адренокортикальная дисплазия. МКБ-10. G31.8 Другие уточнённые дегенеративные болезни нервной системы.
Уильямса синдром (#194050, синдром Уильямса–Бейрена, дефекты генов эластина ELN [130160, 7q11.2], LIM-киназы 1 LIMK [601329] и других [вероятно, протяжённая делеция], Â ). Фенокопии синдрома возможны из-за повышенной чувствительности к витамину D и/или неумеренного приёма эргокальциферола при беременности. Клинически: отставание в умственном развитии, умеренная задержка роста, «лицо эльфа», надклапанный стеноз аорты и/или другие пороки сердца, иногда гиперкальциемия, иногда транзиторный и чаще односторонний парез лицевого нерва.
Ульнарно-маммарный синдром (#181450, 12q24.1, ген TBX3 [601621]). Клинически: дефекты лучевой и локтевой костей, олигодактилия, постаксиальная полидактилия, дефекты малоберцовых костей, задержка роста и полового созревания, ожирение, атрезия заднего прохода, пилоростеноз, врождённый стеноз гортани, паховая грыжа, гипоплазия грудных и апокриновых потовых желёз, патологические зубы, аномалии позвоночника, ДМЖП. Синонимы: синдром Шинцеля, синдром ульнарно-маммарный Паллистера.
Уотсона синдром — наследственное заболевание (193520, ген NF1, VRNF, WSS, дефект гена нейрофибромина 1, 162200, 17q11.2), проявляющееся стенозом лёгочной артерии в сочетании с низкорослостью, пятнами цвета «кофе с молоком», ретроперитонеальными или висцеральными нейрофибромами, ограничением движений в коленном и локтевом суставах, возможны макроцефалия и умственная отсталость
Урофациальный синдром (*236730, синдром Очоа, 10q23–q24, ген UFS, r ). Клинически: специфическое лицо (при смехе лицо принимает выражение кричащего человека), гидронефроз, гидроуретер, крипторхизм, энурез, инфекция мочевого тракта, запоры.
Ухо-нёбно-пальцевой синдром (*311300, Xq28, ген OPD1, À ). Клинически: остеохондродисплазия, сколиоз, патологические локти (углубление в проксимальной части локтевой кости), низкорослость, выступающий лоб, плоское лицо, широкая спинка носа, гипертелоризм, расщелина нёба, гиподонтия, лёгкое снижение слуха, вдавленная грудина, брахидактилия, клинодактилия мизинцев, укорочение ногтей, отсутствие вторичных точек окостенения в крючковатых и головчатых костях запястья; возможно, аналог фронтометафизарной дисплазии. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
ФитцХью–Кёртиса синдром — перигепатит со спайками, сопровождающийся болью в правом подреберье; развивается, как правило, у женщин, перенёсших гонококковый или хламидийный сальпингит.
Фрейзера синдром (136680, 11p13, ген WT1 [194070], Â ). Клинически: дисгенезия и опухоли гонад, первичная аменорея, псевдогермафродитизм, почечная недостаточность.
Фукса синдром. 1. Болезнь неясной этиологии, характеризующаяся множественными буллёзными высыпаниями на коже и слизистых оболочках полости рта, глаз, половых органов, тяжёлым общим состоянием. 2. Гетерохромия осложнённая, Фукса гетерохромия — болезнь неясной этиологии, характеризующаяся дистрофическими изменениями ресничного тела, гетерохромией радужки и развитием катаракты.
Ханта синдром. 1. (*159700, Â ) Миоклонус; интенционный тремор, начинающийся в одной конечности, постепенно усиливающийся и распространяющийся на другие части тела; возможны тоникоклонические судороги; дегенерация зубчатого ядра мозжечка, бледного шара; увеличение содержания мочевой кислоты в ликворе, возможно сочетание с синдромом MERRF « диссинергия мозжечковая миоклоническая Ханта « Рамсэя Ханта синдром. 2. Синдром Рамсея Ханта — паралич лицевого нерва, сочетающийся с герпетическими высыпаниями в наружном слуховом проходе или на коже заушной области. Синонимы: Рамзая Ханта синдром, синдром коленчатого ганглия.
Холла–Ригса синдром (Холла–Ригса умственная отсталость, 234250, r ). Микроцефалия, низкая переносица, вывернутые ноздри, большие губы, скелетная дисплазия, сколиоз, плоские головки и короткие шейки бедренных костей, короткие плечи, плоские эпифизы пальцев рук и лодыжки, задержка роста, умственная отсталость.
Холт–Орама синдром (*142900, 12q24.1, дефект гена TBX5 [601620], Â ) — врождённые аномалии сердца (например, дефекты межпредсердной или межжелудочковой перегородки) в сочетании с различными деформациями предплечья и кисти. Синоним: синдром рука–сердце. МКБ-10. Q87.2 Синдромы врожденных аномалий, вовлекающих преимущественно конечности.
Хэя–Уэллса синдром (106260, Â ): врождённая эктодермальная дисплазия, незначительный гипогидроз, расщепление губы/нёба, жёсткие редкие волосы, рудиментарные веки, дистрофические изменения ногтей, гиподонтия, гипоплазия верхней челюсти. МКБ-10. Q87 Другие уточнённые синдромы врожденных аномалий [пороков развития], затрагивающих несколько систем.
Цефалосиндактилии Грейга синдром (#175700, 7p13, гены GLI3, PAPA, Â ) — мутация онкогена GLI3 (165240). Клинически: полисиндактилия, расщепление большого пальца кисти и стопы, специфическая форма черепа, высокий выступающий лоб, вывих бедра, ускорение костного возраста.
Шварца–Ямпеля–Аберфельда синдром (255800, 1p36.1–p34, ген SJS [SJA], r ) — сочетание миотонии с хондродистрофическими признаками. Клинически: низкий рост, амимичное маленькое лицо, миопия, блефарофимоз, телекант, периодический птоз, микрокорнеа, микрофтальм, юношеская катаракта, множественные ряды ресниц (дистихиаз), остеохондродисплазия, короткая шея, кифосколиоз, деформация грудной клетки («куриная грудь»), пупочная и паховая грыжи, распространённые контрактуры суставов, замедленное моторное развитие, миопатия миотоническая, неразборчивая речь, злокачественная гипертермия. Синонимы: миопатия миотоническая, миотония хондродистрофическая. МКБ-10. Q78.8 Другие уточнённые остеохондродисплазии.
Шёгрена–Ларссона синдром (*270200, 17p11.2, дефекты генов ALDH10, SLS, FALDH, r ) — сочетание гиперкератотических изменений кожи (часто по типу ихтиозиформной эритродермии), отставания в умственном и физическом развитии, спастических параличей, пигментной дегенерации сетчатки, эпилепсии, костных аномалий. Лабораторно: повышение уровня дексадеканола в фибробластах; недостаточность альдегид дегидрогеназы 10. Синоним: недостаточность альдегид дегидрогеназы-10.
Шторморкена синдром (185070, Â ). Клинически: геморрагический диатез, аспления, миоз, мышечная слабость, мигрени, возможен ихтиоз; тельца Хауэлла–Жолли (сферические или овоидные эксцентрично расположенные гранулы; встречаются в циркулирующих эритроцитах, чаще и в большем количестве после спленэктомии).
Эванса синдром — приобретённая аутоиммунная гемолитическая анемия в сочетании с аутоиммунной тромбоцитопенией. Клинически: желтуха, кровоизлияния, экскреция гемосидерина с мочой « Эванса–Фишера синдром. МКБ-10. D69.3 Идиопатическая тромбоцитопеническая пурпура.
Эйди синдром (103100, Â ) — сочетание одностороннего (как правило) замедления зрачковой реакции на конвергенцию, анизокории и изменения величины зрачка в течение дня, отсутствие или ослабление сухожильных рефлексов на нижних (реже на верхних) конечностях вследствие частичной дегенерации нейронов ресничного и спинномозговых узлов. Наблюдают чаще у женщин. После парентерального введения мехолила (неостигмина), как и при семейной дизавтономии, зрачковый и глубокие рефлексы восстанавливаются « Аргайлла Робертсона псевдосиндром « Арефлексия конституциональная « Вейлля–Рея–Эйди синдром « Псевдотабес пупиллотонический « Эйди–Холмса синдром.
Элейалда синдром (акро-цефало-полидактилическая дисплазия, 200995, r ): акроцефальная форма черепа, кавернозные лимфангиомы, постаксиальная полидактилия, кистозная дисплазия почек, полиспления, гипоплазия кишечника, ограничение подвижности диафрагмы.
Элерса–Данло–Русакова синдром — группа наследственных системных заболеваний соединительной ткани (типы и гены: • тип II, 130010, COL5A1, 120215, 9q34.2 q34.3; • тип III, COL3A1, 120180, 2q31; • тип IV, 130050, COL3A1, 120180, 2q31; • тип VI, 225400, PLOD, 153454, 1p36.3 p36.2; • тип VIIA1, 130060, COL1A1, 120150, 17q21.31 q22.05; • тип VIIA2, 130060, COL1A2, 120160, 7q22.1; • тип X (?), FN1, 135600, 2q34; • синдром Элерса Данло Русакова подобный, TNXA, HXBL, TNX, 600261, 6p21.3), вызванных дефектами коллагена (мутации генов коллагеновых полипептидов, недостаточность ферментов модификации коллагенов, например лизин гидроксилазы). Различают минимально 11 генетических разновидностей. Клинически: гиперэластичность и ранимость кожи, разболтанность суставов, травматизация сосудов кожи и крупных артерий. Синонимы: гиперэластическая кожа, эластическая кожа, десмогенез несовершенный, дерматорексис. МКБ-10. Q79.6 Синдром Элерса–Данло.
Эллиса–ван Кревельда синдром (*225500, 4p16, дефект гена EVC, r ) — наследственная хондродисплазия с умственной и физической отсталостью и множественными пороками развития. Синонимы: дисплазия хондроэктодермальная, мезоэктодермальная дисплазия. МКБ-10. Q77.6 Синдром Эллиса–ван Кревельда.
Эпизодической атаксии/миокимии синдром развивается вследствие мутаций гена калиевого канала KCNA1 (176260). Начало: после 10 лет. Клинически: постоянные фасцикуляции, провоцируемые эмоциональными стимулами, сменой позы тела; эпизоды атаксии. Лабораторно: спонтанная активность на электромиограмме, в биоптатах мышц — денервационные изменения.
Эска-Апмарка синдром. Истинная почечная гипоплазия; характерны недоразвитие долек почек, гипертрофия корковой части, глубокие борозды на поверхности.
Якобса синдром (*208250, врождённый семейный гипертрофический синовит, 1q25–q31, ген JCAP, r ). Клинически: артропатия, утренняя скованность, сгибательные контрактуры пальцев, констриктивный перикардит.
Якобсена синдром (147791, 11q23, ген JBS, частичная моносомия 11q, Â ). Клинически: задержка физического и умственного развития, косоглазие, дисморфические черты лица, билатеральная камптодактилия; тромбоцитопения.
Янга синдром — обструктивная азооспермия в сочетании с хроническими инфекциями верхних дыхательных путей (включая синуситы), бронхоэктазы. Сперматогенез, функции ресничного эпителия и содержание фруктозы в сперме не нарушены. МКБ-10. Кодируется в зависимости от состояние, вызвавшее текущее обращение за помощью: J47 Бронхоэктатическая болезнь [бронхоэктаз], N46 Мужское бесплодие.