Синдром миллера дикера что это
Синдром Миллера-Дикера — редкое заболевание неопределенной встречаемости во всех популяциях.
Патогенез синдрома Миллера-Дикера
В регионе делеции синдрома Миллера-Дикера в 17р13.3 картировано более 50 генов, но только ген LIS1 (MIM №601545) связан со специфической фенотипической характеристикой синдрома; гемизиготность по LIS1 вызывает лиссэнцефалию. LIS1 кодирует мозговую изоформу некаталитической бета-субъединицы фактора активации ацетилгидролазы тромбоцитов (PAFAH). PAFAH — ингибитор миграции нейронов и также связывает и стабилизирует микрофибриллы. Предварительные наблюдения указывают, что PAFAH может играть роль в реорганизации микрофибрилл, необходимой для миграции нейронов.
Тем не менее изолированная гаплонедостаточность LIS1 не вызывает других дисморфических признаков, характерных для синдрома Миллера-Дикера. Мутации в гене LIS1 вызывают изолированную лиссэнцефалию (MIM №607432), т.е. лиссэнцефалию без других дисморфии. Поскольку все пациенты с синдромом Миллера-Дикера имеют дисморфические черты лица, этот дисморфизм должен вызываться гаплонедостаточностью одного или нескольких других генов в делеции.
 Синдром Миллера-Дикера
Синдром Миллера-Дикера
Фенотип и развитие синдрома Миллера-Дикера
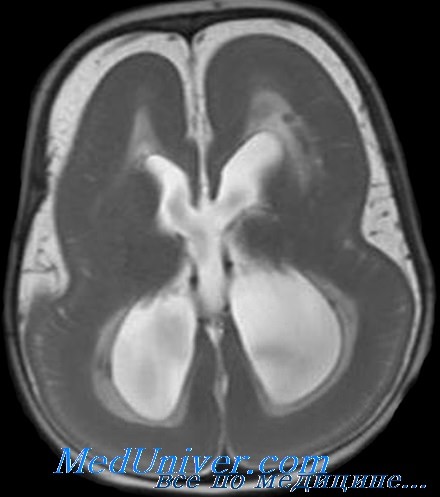
Симптоматика синдрома Миллера-Дикера включает дисгенезию мозга, мышечную гипотонию, задержку развития и лицевые дисморфии. Дисгенезия мозга характеризуется лиссэнцефалией I (полная агирия) или II типа (широко распространенная агирия с несколькими бороздами на лобном или затылочном полюсе), церебральной корой с четырьмя вместо шести слоев, гетеротопией серого вещества и истончением белого вещества. У некоторых больных также отмечают пороки развития сердца и омфалоцеле.
Дети с синдромом Миллера-Дикера плохо едят и растут. Способность улыбаться, краткий визуальный контакт и неспецифические двигательные реакции — единственные способности, приобретаемые большинством пациентов. Кроме умственной отсталости, пораженные обычно страдают от опистотонуса, спастичности и судорог. Почти все они умирают к 2 годам жизни.
Особенности фенотипических проявлений синдрома Миллера-Дикера:
• Возраст начала: пренатальный
• Лиссэнцефалия I или II типа
• Лицевые дисморфии
• Тяжелая общая умственная недостаточность
• Судороги
• Ранняя смерть
 Синдром Миллера-Дикера
Синдром Миллера-Дикера
Лечение синдрома Миллера-Дикера
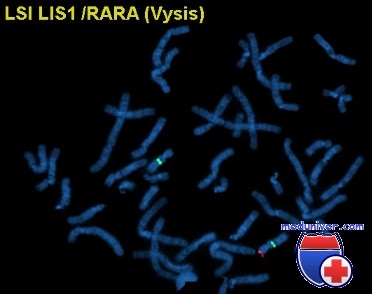
Черты лица больных и обнаружение на МРТ лиссэнцефалии часто позволяет предположить диагноз синдрома Миллера-Дикера. Тем не менее для подтверждения диагноза необходимо обнаружить делецию 17р13.3 при хромосомном анализе или FISH с LIS1-специфическим зондом. Приблизительно 60% пациентов имеют видимую делецию критического региона синдрома Миллера-Дикера.
Синдром Миллера-Дикера неизлечим; следовательно, помощь направлена на коррекцию симптомов и паллиативный уход. Почти всем детям необходимо лекAPCтвенное лечение судорог. Большинство пациентов получают питание через назогастральный или гастростомический зонд из-за проблем вскармливания и повторных аспирации.
Риски наследования синдрома Миллера-Дикера
У 80% пациентов бывает вновь возникшая микроделеция 17р13.3, а 20% наследуют делецию от одного из родителей, несущего сбалансированную хромосомную перестройку. Из-за частоты, с которой деления наследуется от родителя со сбалансированной транслокацией, анализ кариотипа и FISH на LIS1 следует выполнять у обоих родителей. Родитель со сбалансированной транслокацией, захватывающей 17р13.3, имеет приблизительно один шанс из четырех иметь аномального живорожденного ребенка (с синдромом Миллера-Дикера или с дупликацией dupl7p) и приблизительно один шанс из пяти — прерывания беременности. В отличие от этого, если пациент имеет синдром Миллера-Дикера в результате новой делеции, родители имеют низкий риск для повторения синдрома у последующих детей.
Хотя порок развития мозга при синдроме Миллера-Дикера вызван неполной миграцией нейронов в церебральную кору во время 3-4 месяцев гестации, лиссэнцефалию не обнаруживают при МРТ или ультрасонографии плода до конца беременности. Для пренатальной диагностики синдрома Миллера-Дикера необходимо обнаружение делеции 17р13.3 в ворсинах хориона или амнио-цитах плода.
Пример синдрома Миллера-Дикера. Б.Б., мальчик 5 дней жизни, родившийся на 38-й нед гестации, переведен в палату интенсивной терапии отделения новорожденных из-за выраженной гипотонии и затруднений вскармливания. Ребенок родился от неосложненной беременности; УЗИ плода на 14-й нед гестации и материнский сывороточный скрининг на 16-й нед гестации соответствовали норме. Мальчик родился от спонтанных влагалищных родов; оценка по шкале Апгар была 8 баллов на 1-й мин и 9 баллов на 5-й мин жизни.
В семейном анамнезе ребенка генетические, неврологические или врожденные заболевания отсутствуют. При клиническом осмотре выявлены мышечная гипотония и слегка дисморфиче-ские черты лица, включая сужение битемпорального расстояния, вдавленную переносицу, небольшой нос с вывернутыми ноздрями и микрогнатию. В остальном данные обследования в норме. Его анализы на электролиты сыворотки крови, метаболический скрининг и врожденные инфекции оказались в пределах нормы. Ультразвуковое сканирование мозга показало гипоплазию мозолистого тела, легкое расширение желудочков мозга и сглаженную кору.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Миллера-Дикера синдром
OMIM 247200
Наша команда профессионалов ответит на ваши вопросы
Синдром Миллера-Дикера (синдром лиссэнцефалии Миллера-Дикера OMIM 247200) – аутосомно-доминантное заболевание, обусловленное делецией генов локуса 17p13.3 (размер критического региона около 350-400 килобаз). Делеция локуса 17p13.3 приводит к нарушению миграции нейронов и в основном возникает de novo (образуется кольцевая хромосома, либо происходит делеция концевого участка короткого плеча), однако она может наследоваться от родителя со сбалансированной реципрокной транслокацией. Делеции всего гена PAFAH1B1 (другое название LIS1), лежащего в этом локусе, его отдельных экзонов и точковые мутации в нем приводят к развитию изолированной лиссэнцефалии. Формирование специфического фенотипа и более тяжелого течения заболевания при синдроме Миллера-Дикера связано с делецией более протяженного участка хромосомы, захватывающей несколько генов, в частности ген YWHAE, лежащий рядом с геном PAFAH1B1. Описаны случаи делеции только гена YWHAE. При этом у больных формируется фенотип, как при синдроме Миллера-Дикера (задержка роста, черепно-лицевые аномалии, задержка умственного развития), однако наблюдаются иные аномалии развития мозга, среди которых наиболее частыми являются расширение пространств Вирхова-Робина, мальформация Киари I, аномалии мозолистого тела. Описано также несколько случаев дупликации локуса 17p13.3, что также приводило к развитию синдрома Миллера-Дикера. Популяционная частота заболевания неизвестна.
В Центре Молекулярной Генетики методом количественной MLPA проводится поиск делеции / дупликации локуса локуса 17p13.3 и прямое автоматическое секвенирование гена PAFAH1B1.
Публикации в СМИ
Агирия
Агирия (лиссэнцефалия, агирия–пахигирия) — дефект развития в виде слабой выраженности извилин коры головного мозга вследствие нарушения миграции нейробластов в эмбриогенезе. Агирия типа I — основное проявление синдромов Миллера–Дикера и Нормана–Робертса. Агирия типа II (кортикальная дисплазия) — нарушение цитоархитектоники с облитерацией субарахноидального пространства эктопической нейроглиальной тканью и гидроцефалией — компонент синдрома Уокера–Варбурга, врождённой мышечной дистрофии Фукуяма (253800) и некоторых других семейных синдромов.
• Миллера–Дикера синдром агирии (агирия типа I, *247200, три локуса: 17p.13.3, ген LIS1 [601545, в 90% — делеция]; 2p11.2, ген LIS2 [*600217]; второй локус 2q13, r ): микроцефалия, агирия (утолщённая кора с 4 слоями клеток) в сочетании с дефектами развития лицевого скелета и множественными пороками развития внутренних органов.
• Нормана–Робертса синдром — вариант агирии I типа с микроцефалией, низким покатым лбом и выступающей спинкой носа (*257320, r ); в отличие от синдрома Миллера–Дикера, делеции в хр. 17 не обнаружено.
• Уокера–Варбурга синдром (агирия типа II, *236670): сочетание агирии с обструктивной гидроцефалией (микрополигирия).
• Агирия X-сцепленная (*300067, Xq22.3–q23, ген LISX, À доминантное): сочетание агирии с агенезией мозолистого тела.
• Пахигирия с умственной отсталостью и судорожными припадками (600176, r ): тяжёлая энцефалопатия, умственная отсталость, задержка психомоторного развития, генерализованные клоникотонические судороги, утолщение коры мозга до 10–12 мм на МРТ.
МКБ-10. Q04.3 Другие редукционные деформации мозга
Код вставки на сайт
Агирия
Агирия (лиссэнцефалия, агирия–пахигирия) — дефект развития в виде слабой выраженности извилин коры головного мозга вследствие нарушения миграции нейробластов в эмбриогенезе. Агирия типа I — основное проявление синдромов Миллера–Дикера и Нормана–Робертса. Агирия типа II (кортикальная дисплазия) — нарушение цитоархитектоники с облитерацией субарахноидального пространства эктопической нейроглиальной тканью и гидроцефалией — компонент синдрома Уокера–Варбурга, врождённой мышечной дистрофии Фукуяма (253800) и некоторых других семейных синдромов.
• Миллера–Дикера синдром агирии (агирия типа I, *247200, три локуса: 17p.13.3, ген LIS1 [601545, в 90% — делеция]; 2p11.2, ген LIS2 [*600217]; второй локус 2q13, r ): микроцефалия, агирия (утолщённая кора с 4 слоями клеток) в сочетании с дефектами развития лицевого скелета и множественными пороками развития внутренних органов.
• Нормана–Робертса синдром — вариант агирии I типа с микроцефалией, низким покатым лбом и выступающей спинкой носа (*257320, r ); в отличие от синдрома Миллера–Дикера, делеции в хр. 17 не обнаружено.
• Уокера–Варбурга синдром (агирия типа II, *236670): сочетание агирии с обструктивной гидроцефалией (микрополигирия).
• Агирия X-сцепленная (*300067, Xq22.3–q23, ген LISX, À доминантное): сочетание агирии с агенезией мозолистого тела.
• Пахигирия с умственной отсталостью и судорожными припадками (600176, r ): тяжёлая энцефалопатия, умственная отсталость, задержка психомоторного развития, генерализованные клоникотонические судороги, утолщение коры мозга до 10–12 мм на МРТ.
МКБ-10. Q04.3 Другие редукционные деформации мозга
Лиссэнцефалия
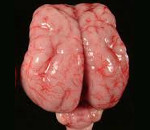
Лиссэнцефалия – группа генетически обусловленных аномалий развития головного мозга, характеризующихся частичным или полным недоразвитием извилин и борозд коры больших полушарий, а также нарушением ее ультраструктуры. Выраженность и сочетания симптомов этого состояния различаются при разных формах заболевания, наиболее распространены судороги, глубокая умственная отсталость, нарушения глотания и гипотония мышц. Диагностика лиссэнцефалии может производиться ультразвуковыми методиками (в том числе и пренатально), компьютерной и магнитно-резонансной томографией, для наиболее распространенных форм возможно определение посредством молекулярно-генетического анализа. Специфического лечения не существует, используют симптоматическую и поддерживающую терапию.
Общие сведения
Лиссэнцефалия – группа тяжелых аномалий развития головного мозга, которые сопровождаются недоразвитием коры больших полушарий с формированием пахигирии (наличием всего нескольких извилин и борозд) или агирии (полным отсутствием складчатости коры). Данная патология может выступать в качестве самостоятельного генетического заболевания или входить в симптомокомплекс других синдромов – например, Миллера-Дикера, Фукуямы и Уокера-Варбурга. Механизм наследования различных типов лиссэнцефалии может быть аутосомно-рецессивным, аутосомно-доминантным (в данном случае чаще всего имеют место спонтанные или герминативные мутации) и сцепленным с Х-хромосомой. Из-за многообразия механизмов наследования половое распределение нарушения неодинаково при различных формах патологии. Лиссэнцефалия является достаточно редкой генетической аномалией развития мозга, поэтому встречаемость определена только для наиболее распространенной первой группы нарушений – она составляет 11,7 случаев на 1 000 000 новорожденных. Для остальных групп лиссэнцефалии встречаемость не установлена, в том числе и потому, что во многих случаях плод с подобной патологией не вынашивается, и беременность самопроизвольно прерывается в первом триместре еще до определения наличия порока.
Причины лиссэнцефалии
Основная общая причина всех типов лиссэнцефалии – нарушение процесса миграции клеток-предшественников нейронов (нейробластов) из передних отделов нервной трубки к будущей коре больших полушарий. В результате этого вместо сложной складчатой структуры, которая имеет в своем составе шесть слоев, образуется гладкая или имеющая в разы меньше борозд кора, состоящая из 2-4 слоев (в зависимости от формы заболевания). Поскольку кора больших полушарий у человека отвечает за когнитивные функции, в ней содержится огромное число нервных центров и обширные ассоциативные зоны, лиссэнцефалия приводит к тяжелейшим расстройствам. Кроме того, при некоторых типах мутаций, вызывающих данное состояние, возможно развитие аномалий других органов и тканей, что еще больше усугубляет состояние больного.
Наиболее распространенные типы лиссэнцефалии обусловлены дефектами гена PAFAH1B1, также известного под названием LIS1 и расположенного на 17-й хромосоме. Многие врачи-генетики отмечают, что для возникновения выраженного недоразвития коры головного мозга и синдрома Миллера-Дикера необходимы не точечные мутации в LIS1, а крупные делеции с захватом сотен пар азотистых оснований. Нередко при этом повреждаются и окружающие гены, что становится причиной разнообразных фенотипических проявлений данного типа лиссэнцефалии. Ген LIS1 кодирует внутриклеточную субъединицу сложного по своей структуре фермента, который принимает активное участие в миграции нейробластов. Дефекты в структуре этой субъединицы приводят к нарушению данного процесса и развитию данного заболевания.
Другая форма лиссэнцефалии обусловлена мутацией гена DCX, локализованного на Х-хромосоме, поэтому наследование этого типа патологии сцеплено с полом. Белок, получаемый в результате экспрессии данного гена, участвует в формировании особого типа микротрубочек, которые вырабатываются только нейробластами и необходимы им для формирования связей между клетками. Нарушения в структуре DCX приводят к синтезу дефектного белка, что провоцирует лиссэнцефалию. Другой относительно изученный вариант этого состояния, также сцепленный с Х-хромосомой, вызывается мутациями гена ARX, который является фактором транскрипции для иных генов. Он контролирует развитие коры больших полушарий, поджелудочной железы и половых органов, из-за чего дефекты ARX проявляются многочисленными пороками данных органов, в том числе лиссэнцефалией.
Еще одним распространенным вариантом лиссэнцефалии является форма заболевания, обусловленная мутацией гена RELN, расположенного на 7-й хромосоме. Дефекты этого гена приводят к так называемому синдрому Норман-Робертса, который, помимо прочего, сопровождается выраженным нарушением складчатости коры больших полушарий. Ген RELN кодирует последовательность гликопротеида рилина, который принимает активное участие в формировании нервной ткани, а также контролирует образование новых дендритов и функционирование долговременной памяти у взрослых людей. Удалось идентифицировать еще один ген, приводящий к развитию лиссэнцефалии – TUBA1A, который локализован на 12-й хромосоме. Он кодирует определенный компонент цитоскелета нейронов, его дефект приводит к неполноценности нейробластов, что нарушает процесс их миграции в эмбриональном периоде.
Классификация лиссэнцефалии
На сегодняшний день выявлено более двадцати вариантов лиссэнцефалии. Эти варианты обусловлены различными мутациями, имеют отличия в фенотипических проявлениях заболевания, разные механизмы наследования и тяжесть симптомов. Долгое время разновидности патологии не удавалось успешно классифицировать из-за значительного разнообразия генетических дефектов и недостаточных данных о ключевых генах, приводящих к развитию некоторых форм заболевания. Лишь в 2003-м году удалось создать достаточно приемлемую с точки зрения современной генетики и неврологии классификацию лиссэнцефалии, которая учитывает основные нюансы этого состояния. Специалисты разделяют все формы лиссэнцефалии на пять классов:
Данная классификация критикуется некоторыми исследователями по причине наличия большого количества «белых пятен» в виде включения в нее форм лиссэнцефалии с неопределенными ключевыми генами. Однако в настоящее время именно это разделение считается наиболее общепринятым в научном мире. Классификация корректируется по мере дальнейшего изучения данного состояния.
Симптомы лиссэнцефалии
Основными проявлениями лиссэнцефалии являются мышечная слабость, выявляемая уже при рождении, частые расстройства глотания и сосания, судорожные припадки, сильное отставание в физическом и умственном развитии. Выраженность тех или иных проявлений заболевания зависит от наличия пороков развития как головного мозга, так и других органов и систем, а также от степени недоразвития коры. Наиболее тяжелые формы патологии наблюдаются при наличии полной агирии. Иногда больные лиссэнцефалией могут доживать до подросткового и даже взрослого возраста, глубокое недоразвитие центральной нервной системы сохраняется в течение всей жизни. Некоторые пациенты проживают жизнь на грани вегетативного состояния. Летальный исход при лиссэнцефалии наступает из-за осложнений, обусловленных пороками других органов и систем, а также из-за вторичных пневмоний, сердечно-сосудистых и иных аномалий.
Диагностика и лечение лиссэнцефалии
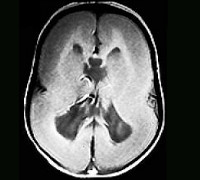
В отношении лиссэнцефалии возможна пренатальная диагностика при помощи ультразвуковых методов исследования, при этом увеличение разрешающей способности УЗИ-аппаратуры способствует все более раннему определению заболевания. Развитие нарушений в строении коры больших полушарий происходит на 14-20-й неделе гестации, в настоящее время уже в этот период можно определить патологию и поставить вопрос о прерывании беременности. После рождения ребенка с подозрением на лиссэнцефалию диагноз подтверждают при помощи КТ и МРТ. Молекулярно-генетическая диагностика обладает высокой точностью, но она доступна только в отношении тех форм заболевания, для которых известны ключевые гены. Специфического лечения лиссэнцефалии не существует, применяют противосудорожные препараты, ноотропы и другие средства, позволяющие уменьшить выраженность симптоматики. При наличии иных пороков развития производится их коррекция по медицинским показаниям.
Прогноз и профилактика лиссэнцефалии
Прогноз практически любой формы лиссэнцефалии крайне неблагоприятный, большинство больных умирает в раннем детстве от осложнений и других пороков развития. Описаны отдельные легкие случаи этого состояния с частичным или очаговым недоразвитием коры больших полушарий, но не все специалисты склонны причислять такие типы патологии к лиссэнцефалиям. Профилактика возможна только в рамках пренатальной диагностики. При наличии в роду подобных заболеваний имеет смысл провести генетический анализ на носительство генов аутосомно-рецессивных типов патологии. При обнаружении у плода нарушений формирования мозга, соответствующих лиссэнцефалии, ставится вопрос о прерывании беременности.
СОДЕРЖАНИЕ
Презентация
Мозг необычно гладкий, с меньшим количеством складок и бороздок. Лицо, особенно у детей, имеет отличительные характеристики, включая короткий нос с вздернутыми ноздрями, утолщенную верхнюю губу с тонкой красной верхней границей, выступающие лобные выступы, небольшую челюсть, низко посаженные назад повернутые кзади уши, запавшее лицо в середине лица, широко расставленные глаза и гипертелоризм. Лоб выпуклый, с прикусной впадиной.
Причина
Диагностика
Заболевание можно диагностировать с помощью цитогенетических методов, таких как флуоресцентная гибридизация in situ (FISH), тестирование микроделеции в LIS1.
Раннее обнаружение
С помощью пренатальной ультрасонографии можно увидеть раннее выявление аномалий развития мозга у плода с МДС. При рождении у младенца может присутствовать лицевой дисморфизм. Маленькие дети, когда они поражены, могут страдать от проблем с кормлением, серьезной умственной отсталости, задержки развития и судорог. МРТ облегчает раннее выявление этого синдрома у детей, выявляя изображение «гладкого мозга», также называемое лиссэнцефалией. Дети с этим синдромом могут оставаться недиагностированными из-за редкости и преобладания дисморфических черт лица. Синдром имеет общие внешние признаки ( фенотип ), аналогичные более распространенным синдромам. Отсутствие соответствующего семейного анамнеза может задержать постановку диагноза. FDNA предоставляет услугу, которая, в свою очередь, увеличивает шансы обнаружения этих отличительных характеристик, которые при показе генетику могут помочь в постановке правильного медицинского диагноза. Если у пары был один ребенок с МДС, им могут предложить пренатальный скрининг на будущие беременности. Этот вариант особенно важен для 20% семей с МДС, в которых один из родителей несет сбалансированную хромосомную перестройку. Риск рождения у этих пар еще одного ребенка с МДС зависит от конкретного типа хромосомной перестройки и может достигать 25–33%. Для семей, в которых хромосомы обоих родителей нормальные, риск рождения еще одного ребенка с МДС невелик (1% или меньше). На ранних сроках беременности можно использовать отбор проб ворсинок хориона (CVS) или амниоцентез для получения небольшого образца клеток развивающегося эмбриона для хромосомных исследований. Ранняя пренатальная диагностика с помощью ультразвука не является надежной, потому что мозг обычно остается гладким до более поздних сроков беременности. Пары, которые рассматривают возможность пренатальной диагностики, должны обсудить риски и преимущества этого типа тестирования с генетиком или генетическим консультантом.
Визуализация мозга
Хотя лекарства от МДС пока нет, многие осложнения, связанные с этим состоянием, можно вылечить, и многое можно сделать для поддержки или компенсации функциональных нарушений. Из-за разнообразия симптомов может потребоваться обращение к разным специалистам и прохождение различных обследований, в том числе:
Это заболевание встречается довольно редко, и лечения не хватает.
Прогноз
Большинство людей с этим заболеванием не выживают после детства. Люди с МДС обычно умирают в младенчестве и поэтому не доживают до того возраста, когда они могут воспроизвести и передать МДС своему потомству. Благодаря более точной диагностике с помощью МРТ и улучшенному лечению симптомов с помощью медицинских методов лечения, включая гастростомию (питательные трубки), аспирационные аппараты, противосудорожные препараты, трахеостомию и управление выделениями, больше детей доживают до взрослого возраста и имеют лучшее качество жизни. Развитие навыков и основной силы можно улучшить с помощью физиотерапии, но улучшение ограничено тяжестью состояния. Некоторые дети могут сидеть, ползать или произносить несколько простых слов, но у большинства из них наблюдается серьезная задержка в развитии, и они остаются на уровне младенцев на всю жизнь. В любом возрасте наиболее частой причиной смерти является пневмония, часто после того, как несколько случаев привели к ослаблению дыхательной функции.
Эпидемиология
Миллер-Дикер встречается менее чем у одного человека из 100 000 и может встречаться у всех рас. Учитывая эту статистику, можно сделать вывод, что около 75000 человек во всем мире страдают от МДС.
История
MDS был назван в честь двух врачей, Джеймса К. Миллера и Х. Дикера, которые независимо друг от друга описали это состояние в 1960-х годах. Отличительным признаком МДС является лиссэнцефалия, состояние, при котором внешний слой мозга, кора головного мозга, является аномально толстым и не имеет нормальных извилин (извилин). В некоторых областях мозга извилин меньше, но они шире, чем обычно (пахигири). В других местах извилины (агири) отсутствуют. Обычно в течение третьего и четвертого месяцев беременности клетки мозга ребенка размножаются и перемещаются на поверхность мозга, образуя кору. Лиссэнцефалия вызывается нарушением миграции нервных клеток. МДС часто называют лиссэнцефалией Миллера-Дикера.
Дж. К. Миллер описал это заболевание, а в 1969 г. Х. Дикер подчеркнул, что ему следует также назвать синдром лиссэнцефалии, потому что несколько пороков развития возникают за пределами самого мозга. Когда впервые был описан МДС, генетики предположили, что он следует аутосомно-рецессивному типу наследования. В начале 1990-х годов у нескольких пациентов с синдромом Миллера-Дикера было обнаружено отсутствие небольшой части хромосомы 17. (17p13.3) (частичная делеция).