Синдром Лея

Синдром Лея (СЛ) — это гетерогенное генетически обусловленное заболевание, относящееся к группе митохондриальных энцефаломиопатий с аутосомно-рецессивным или митохондриальным типом наследования и связанное с мутациями в генах, кодирующих полипептиды комплексов дыхательной цепи митохондрий, а также белков, принимающих участие в их сборке на внутренней поверхности митохондриальной мембраны [4].
Первые описания данного синдрома были даны Денисом Леем в 1951 году [1].
На заре открытия данного заболевания появились предположения, что его причиной является нарушение метаболизма, однако они не получили своего клинического подтверждения.
Доктор F. Hommes в 1968 году описал семьи, у представителей которых наблюдалось снижение активности пируваткарбоксилазы [2]. С 1975 года появились данные, что причиной СЛ может являться недостаточность пируватдегидрогеназного комплекса, а позже были обнаружены молекулярно-генетические нарушения. С развитием генетики и биохимии было выяснено, что изменения генов в мтДНК, которые ответственны за кодирование субъединицы АТФ-азы или тРНК — ядерных генов, кодирующих полипептиды комплекса дыхательной цепи митохондрий, — а также за нарушения в генах, отвечающих за сборку КДЦМ на митохондриальной мембране, приводят к развитию синдрома Лея [3]. Всего насчитывается 12 генов: NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, NDUFV1, SDHA, SURF1, COX10, COX15, SCO2, BCS1L [1].
Чаще всего дефект обнаруживается в результате недостаточности IV КДЦМ-цитохром С-оксидазе (COX). Этот фермент является последним в электронно-транспортной системе митохондрий. СОХ включает в себя 13 субъединиц: 11 кодируются ядерными генами, 3 – мтДНК.
Чаще всего данная патология возникает в результате мутации гена SURF1, который лежит в хромосоме 9q34, кластере 6. Данный белок включен во внутреннюю мембрану митохондрии, и нарушения в нем приводят к синтезу укороченного белка и, как следствие, к повреждению СОХ-комплекса [4].
В результате биохимических исследований у большинства пациентов с СЛ обнаруживается повышение лактата в крови и спинномозговой жидкости. Повышение соотношения лактат/пируват является отражением нарушения окислительно-восстановительного баланса в цитоплазме [4].
Те пациенты, у которых мутации не превышают 70% от всех мтДНК к конкретной ткани, не имеют клинических проявлений данного синдрома. Однако при превышении данного порога могут проявляться симптомы, которые будут описаны ниже [1].
Синдром Лея чаще всего проявляется в детстве в виде утраты уже имеющихся психомоторных навыков, мозжечковыми и экстрапирамидными расстройствами, судорогами, мышечной гипотонией. Однако могут проявляться и такие симптомы как нистагм, потеря слуха, атрофия зрительного нерва. Дети отстают в своем развитии, отмечается регресс психомоторных функций, вялость, сонливость. Такие пациенты склонны к лактоацидозам. Ночью и при физических нагрузках возникают проблемы с дыханием в виде апноэ, диспноэ, тахипноэ [1].
При МРТ-диагностике наблюдается снижение МР-сигнала в различных областях ГМ.
Также пациентам с синдромом Лея свойственна «парадоксальная гиперкетонемия» — повышение уровня кетоновых тел после пищевой нагрузки и высокое соотношение 3-гидроксибутират/ацетоацетат в крови. При проведении анализа органических кислот мочи может наблюдаться повышенная экскреция органических кислот, участвующих в цикле Кребса (фумаровая, янтарная и др.) [4].
Синдром Лея имеет следующую классификацию:
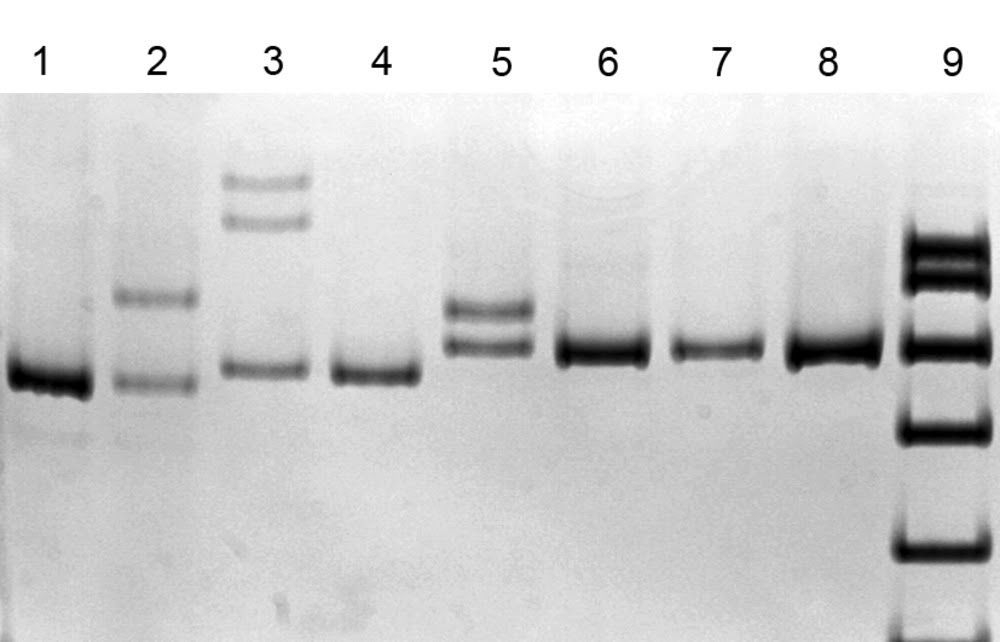
ДНК-диагностика 3 частых мутаций в гене SURF1 с помощью метода SSCP анализа (электрофорез в 8% полиакриламидном геле) [4].
.
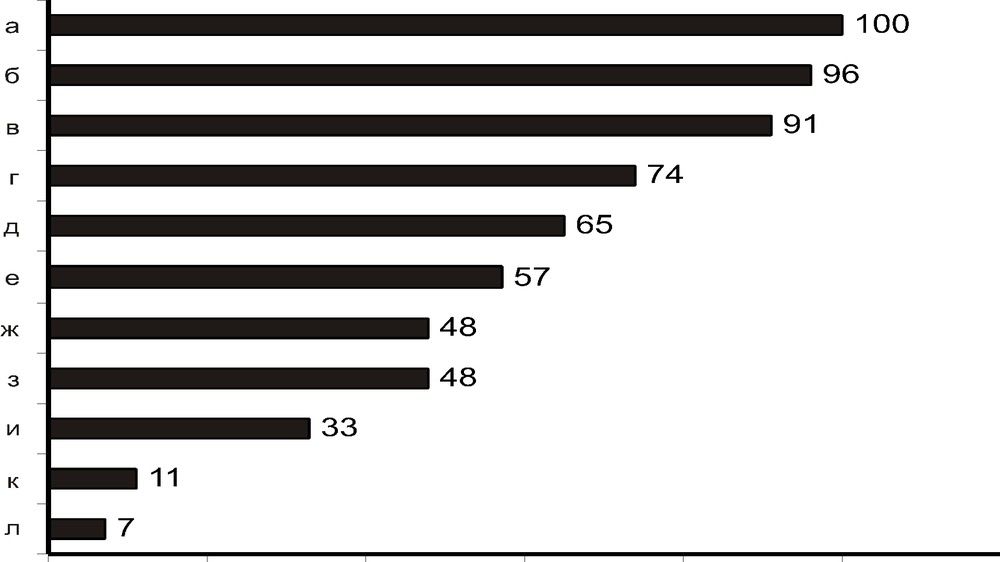
Статистика основных синдромов [4]:
Источники:
Синдром Лея (болезнь Лея)

Общие сведения
Синдром Лея относится к редким нейрометаболическим синдромам, поражающим ЦНС, и вызывающим нарушение координации движений и мышления и летальный исход. Заболевание наследственное по аутосомно-рецессивному типу или Х-сцепленному. Патологии чаще всего подвержены маленькие детки до 2-х лет – 1-2 ребенка на десятки тысяч, в более редких случаях – подростки и взрослые особы. Код по МКБ-10: G 31.8
Миру стало известно об этом заболевании благодаря Денису Лею, который описал его еще в 1951 году. В научных кругах обособить синдром от подобных энцефалопатий удалось в 1954 г. Связь с митохондриальной активностью смогли выявить 1968 г, но главным продвижением стало обнаружение мутации в цитохромоксидазах в 1977 г.
Проблема состоит в том, что эффективных препаратов или методов лечения болезни Лея до сих пор не разработано, ведь причин её множество, начиная с влияния мутаций в генах, отвечающих за работу митохондрий, и заканчивая – осложнениями дегенеративных заболеваний нервной системы, аномальными образованиями некротизированных очагов или прорастания сосудов, глиоза в структурах головного мозга.
Единственным профилактическим методом на сегодня является «рождение детей от трех родителей», то есть благодаря использованию донорских яйцеклеток и митохондриальной донации.
Патогенез
Для таких митохондриальных заболеваний как синдром Лея характерно полиорганное поражение с вовлечением нервных и мышечных тканей. В основе патологии обычно лежит сбой регулирования обмена пировиноградной кислоты, при этом наблюдается нарушение обменных реакций, снижение выработки энергии и замедление процессов перемещения электронов в клетках дыхательной системы. Процессы могут спорадически усилиться, чему способствует наследственные факторы. Для запуска развития синдрома необходимо присутствие в организме более 90% мутантной мтДНК от всей мтДНК. В случае меньшего содержания мутантной ДНК симптоматика напоминает нейропатию и сводится к атаксии и пигментному ретиниту.
Для болезни характерно раннее развитие и стремительное злокачественное течение с присоединением большого количества различных неврологических нарушений, затем начинаются проблемы с дыханием и метаболизмом.
Классификация
В зависимости от патогенеза и возрастных особенностей выделяют различные варианты нарушений, выражающиеся в виде:
В зависимости от течения синдром Лея бывает:
Причины
Главным провоцирующим фактором синдрома Лея принято считать недостаточность цитохромоксидазы и мутации генов, отвечающих за работу митохондрий:
Однако, способствовать нейрометаболической энцефалопатии может:
Симптомы
Синдром Лея и его прогрессирование приводит:
Клиническая характеристика синдрома Лея при других формах патологии
Клиническая характеристика и патогенез схожи, но симптоматика может дополняться:
Анализы и диагностика
Болезнь Лея относится к подострым либо хроническим некротизирующим энцефаломиопатиям и является важным направлением изучения в детской невралгии. Для её диагностики необходима консультация у врача-невролога, а также:
Лечение
Медикаментозное консервативное лечение болезни Лея обычно симптоматическое и включает использование:
Синдром Ли (Лея)
Внимание! Информация носит ознакомительный характер и не предназначена для постановки диагноза и назначения лечения. Всегда консультируйтесь с профильным врачом!
Что такое синдром Ли?
Синдром Ли (синдром Лея) — это редкое генетическое нейрометаболическое заболевание. Он характеризуется дегенерацией центральной нервной системы (т.е. головного мозга, спинного мозга и зрительного нерва). Симптомы синдрома Ли обычно начинаются в возрасте от трех месяцев до двух лет, но у некоторых пациентов признаки и симптомы проявляются лишь через несколько лет.
Симптомы связаны с прогрессирующим неврологическим ухудшением состояния и могут включать потерю ранее приобретенных двигательных навыков, потерю аппетита, рвоту, раздражительность и/или судорожную активность. По мере прогрессирования синдрома Лея симптомы могут также включать генерализованную слабость, отсутствие мышечного тонуса (гипотонию) и эпизоды лактоацидоза, которые могут привести к нарушению дыхательной и почечной функции.
Синдром Лея может быть вызван несколькими различными генетически обусловленными дефектами ферментов, первоначально описанными более 60 лет назад. Большинство людей с синдромом Ли имеют дефекты производства митохондриальной энергии, такие как дефицит фермента комплекса дыхательной цепи митохондрий или пируватдегидрогеназного комплекса. В большинстве случаев синдром Ли наследуется по аутосомно-рецессивному типу. Тем не менее, Х-сцепленное рецессивное и наследование по материнской линии вследствие мутации митохондриальной ДНК являются дополнительными путями передачи.
Признаки и симптомы
Симптомы классического синдрома Ли (инфантильная некротическая энцефалопатия), быстро прогрессирующее неврологическое расстройство, обычно начинаются в возрасте от 3 месяцев до 2 лет. У большинства детей первым заметным признаком является потеря ранее приобретенных двигательных навыков. Когда у болезни раннее начало (т.е., 3 месяца), потеря контроля над головой и плохая способность сосать могут быть первыми заметными симптомами. Она может сопровождаться глубокой потерей аппетита, периодической рвотой, раздражительностью, постоянным плачем и возможными приступами судорог. Задержки в достижении основных этапов развития также могут возникнуть. Пострадавшие дети могут не расти и набирать вес с ожидаемой скоростью (неспособность развиваться).
Если синдром Ли проявляется позже в детстве (например, через 24 месяца), ребенок может испытывать трудности с формулированием слов (дизартрия) и координацией произвольных движений, таких как ходьба или бег (атаксия). Ранее приобретенные интеллектуальные навыки могут уменьшиться, и интеллектуальная инвалидность также может возникнуть.
Прогрессирующее неврологическое ухудшение, связанное с синдромом Лея, характеризуется различными симптомами, включая:
Дальнейшее неврологическое развитие задерживается.
Могут возникнуть эпизоды лактоацидоза, которые характеризуются аномально высоким уровнем молочной кислоты в крови, мозге и других тканях организма. Периодически уровень углекислого газа в крови также может быть ненормально повышен (гиперкапния). Лактоацидоз и гиперкапния могут привести к психомоторной регрессии и нарушениям дыхания, сердца или почек.
У детей с синдромом Ли обычно возникают проблемы с дыханием, включая временное прекращение самопроизвольного дыхания (апноэ), затрудненное дыхание (одышка), ненормально быстрое дыхание (гипервентиляция) и/или патологическое дыхание Чейна-Стокса. У некоторых детей могут также возникнуть трудности с глотанием (дисфагия). Проблемы со зрением могут включать ненормально быстрые движения глаз (нистагм), вялые зрачки, косоглазие, паралич определенных глазных мышц (офтальмоплегия), ухудшение нервов глаз (зрительная атрофия) и/или нарушение зрения, приводящее к слепоте.
Синдром Ли также может повлиять на сердце. У некоторых детей с этим расстройством может наблюдаться аномальное расширение сердца (гипертрофическая кардиомиопатия) и разрастание фиброзной мембраны, которая разделяет различные камеры сердца (асимметричная септальная гипертрофия). Со временем может возникнуть заболевание, поражающее нервы вне центральной нервной системы (периферическая нейропатия), вызывающее прогрессирующую слабость рук и ног.
Симптомы Х-связанной инфантильной формы синдрома Лея схожи с симптомами классического синдрома Лея. Симптомы возникновения синдрома Ли у взрослых (подострая некротическая энцефаломиелопатия), очень редкая форма расстройства, обычно начинаются в подростковом или раннем взрослом возрасте. Начальные симптомы, как правило, связаны со зрением и могут включать такие аномалии, как размытые центрального поля зрения (центральная скотома), дальтонизм и/или прогрессирующую потерю зрения из-за дегенерации зрительного нерва (двусторонняя зрительная атрофия). При этой форме расстройства неврологические проблемы, связанные с заболеванием, прогрессируют медленно. Приблизительно в 50 лет больным людям становится все труднее координировать произвольные движения (атаксия).
Дополнительные поздние симптомы могут включать частичный паралич и непроизвольные мышечные движения (спастический парез), внезапные мышечные спазмы (клонические подёргивания), генерализованные тонико-клонические судороги и/или деменцию различной степени тяжести.
Причины
Несколько различных типов генетически детерминированных метаболических дефектов могут привести к синдрому Ли. Состояние может быть вызвано дефицитом одного или нескольких различных ферментов (например, ферментов митохондриальной дыхательной цепи или ферментных компонентов комплекса пируватдегидрогеназы). Эти недостатки фермента вызваны изменениями (мутациями) в одном из нескольких различных генов заболевания (генетическая гетерогенность). Эти мутации могут быть унаследованы как аутосомно-рецессивный признак, Х-связанный рецессивный признак или как мутация, обнаруженная в ДНК митохондрий. В некоторых случаях синдрома Лея генетическая причина не может быть идентифицирована.
Генетическая информация содержится в двух типах ДНК: Нуклеарный ДНК (нДНК) содержится в ядре клетки и наследуется от обоих биологических родителей. Митохондриальная ДНК (мтДНК) содержится в митохондриях клеток и наследуется исключительно от матери ребенка. Генетические заболевания, обусловленные мутациями нДНК (изменение генетического материала), определяются двумя генами, один из которых получен от отца, а другой — от матери. Рецессивные генетические нарушения возникают, когда человек наследует один и тот же аномальный ген по одному признаку от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно бессимптомным. Риск того, что двое родителей-носителей оба передадут дефектный ген и, следовательно, заразят ребенка, составляет 25% при каждой беременности. Риск родить ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность для ребенка получить нормальные гены от обоих родителей и быть генетически нормальным для этой конкретной черты составляет 25%.
Другие дефициты энзимов на основе нДНК (например, NADH-CoQ и цитохром с-оксидаза) также являются причиной некоторых случаев аутосомно-рецессивного синдрома Ли. Эти специфические недостатки фермента были связаны с несколькими различными генами. Например, мутации гена SURF1, расположенного на хромосоме 9, вызывают синдром Ли, связанный с дефицитом цитохром с-оксидазы. Все эти различные генетические дефекты, по-видимому, имеют общее влияние на центральную нервную систему, что приводит к прогрессирующему неврологическому ухудшению.
В медицинской литературе также имеются данные о рецессивной форме синдрома Лея, сцепленной с Х-хромосомой нДНК. Эта форма заболевания была связана со специфическим дефектом гена, известного как E1-альфа-субъединица пируватдегидрогеназного комплекса, расположенного на короткой руке (p) Х-хромосомы (Xp22.2-22.1). Х-связанные рецессивные расстройства представляют собой состояния, которые кодируются на Х-хромосоме. У женщин две Х-хромосомы, а у мужчин одна Х-хромосома и одна Y-хромосома. Следовательно, у женщин признаки заболевания на Х-хромосоме могут маскироваться нормальным геном на другой Х-хромосоме. Поскольку у мужчин есть только одна Х-хромосома, если они наследуют ген заболевания, присутствующего на Х, оно будет выражено. Мужчины с Х-связанными нарушениями передают ген всем своим дочерям, которые будут носительницами заболевания, но никогда сыновьям. Женщины, являющиеся носительницами Х-связанного расстройства, имеют 50-процентный риск передачи состояния носителя своим дочерям и 50-процентный риск передачи заболевания своим сыновьям.
В некоторых случаях синдром Ли может быть унаследован от матери как мутация, обнаруженная в ДНК митохондрий. Митохондрии, найденные сотнями или тысячами практически в каждой клетке тела, регулируют выработку клеточной энергии и несут генетические чертежи этого процесса в своей собственной уникальной ДНК (мтДНК). МтДНК от отца несут сперматозоиды. Однако в процессе оплодотворения мтДНК отца теряется. В результате вся человеческая мтДНК происходит от матери. Пострадавшая мать передаст черты всем своим детям, но только дочери передадут мутацию(и) следующему поколению.
Генетические мутации, которые присутствуют в мтДНК, могут превосходить по численности нормальные копии генов. Симптомы могут не проявляться до тех пор, пока мутации не появятся в значительном проценте митохондрий. Неравномерное распределение нормальной и мутантной мтДНК в разных тканях организма может повлиять на разные системы органов у людей из одной семьи и может привести к различным симптомам у затронутых членов одной семьи.
Специфический дефект мтДНК, который может быть ответственным за некоторые случаи синдрома Лея (мтДНК nt 8993), связан с геном, известным как АТФаза 6 (дефицит V комплекса митохондриальной дыхательной цепи [дефицит АТФазы]). Эти случаи иногда называют наследственным синдромом Ли или синдромом Лея, связанным с мтДНК.
Некоторые исследователи считают, что случаи синдрома Ли у взрослых могут быть унаследованы как аутосомно-доминантный признак вследствие мутации нДНК. Доминантные генетические нарушения возникают, когда для появления заболевания необходима только одна копия ненормального гена. Поскольку это состояние связано с мутацией нДНК, аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации нДНК у пострадавшего человека. Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50 процентов при каждой беременности, независимо от пола ребенка.
Затронутые группы населения
Классическая форма синдрома Ли развивается в младенчестве (детская некротизирующая энцефалопатия) и обычно начинается в возрасте от 3 месяцев до 2 лет. Эта форма заболевания поражает мужчин и женщин в равных количествах.
В случаях синдрома Ли, которые наследуются как рецессивный признак, связанный с Х, симптомы обычно развиваются в младенчестве. Эта форма заболевания поражает почти вдвое больше мужчин, чем женщин.
В некоторых редких случаях синдром Лея может начаться в позднем подростковом или раннем взрослом возрасте (подострая некротическая энцефаломиелопатия у взрослых). В этих случаях, которые поражают вдвое больше мужчин, чем женщин, прогрессирование заболевания происходит медленнее, чем классическая форма заболевания.
Исследователи когда-то полагали, что классическая форма синдрома Ли приходится примерно в 80 процентах случаев. В медицинской литературе распространенность синдрома Ли оценивается в 1 на 36 000–40 000 живорождений.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Ли. Сравнения могут быть полезны для дифференциальной диагностики:
Диагностика
Диагноз синдрома Лея может быть подтвержден тщательной клинической оценкой и различными специализированными тестами, в частности передовыми методами визуализации. Сканирование мозга с помощью магнитно-резонансной томографии (МРТ) или компьютерной томографии (КТ) может выявить аномальные участки в определенных частях мозга (т.е. базальных ганглиях, стволе мозга и сером веществе). МРТ использует магнитное поле и радиоволны для получения изображений поперечных сечений отдельных органов и тканей организма. Во время КТ-сканирования компьютер и рентген используются для создания пленки, показывающей изображения в поперечном разрезе определенных структур ткани. В коре головного мозга могут обнаружится маленькие или большие кисты.
Лабораторные тесты могут выявить высокий уровень кислых отходов в крови (лактоацидоз), а также повышенный уровень пирувата и аланина. Сахар в крови (глюкоза) может быть немного ниже, чем обычно. Фермент пируваткарбоксилаза может отсутствовать в печени, а в крови и моче больных может присутствовать ингибитор продукции тиаминдифосфата. У некоторых детей с синдромом Ли может обнаруживаться дефицит ферментов пируватдегидрогеназного комплекса или цитохром с-оксидазы.
Стандартные методы лечения
Не существует проверенных методов лечения синдрома Ли любого типа. Рекомендации по лечению основаны главным образом на открытых исследованиях, отчетах о случаях и личных наблюдениях. Лечение синдрома Ли направлено на конкретные симптомы, которые проявляются у каждого человека. Лечение может потребовать скоординированных усилий команды специалистов. Педиатрам, кардиологам, неврологам, специалистам по оценке и лечению проблем со слухом (аудиологам), глазным специалистам и другим медицинским работникам может потребоваться систематическое и всестороннее планирование эффективного лечения ребенка.
Наиболее распространенным методом лечения синдрома Лея является введение тиамина (витамина В1) или его производных. У некоторых людей с этим расстройством может наблюдаться временное симптоматическое улучшение и небольшое замедление прогрессирования заболевания. Тем пациентам с синдромом Ли, у которых также наблюдается дефицит ферментного комплекса пируватдегидрогеназы, может быть рекомендована диета с высоким содержанием жиров и низким содержанием углеводов.
Прогноз
Прогноз синдрома Ли плохой, ожидаемая продолжительность жизни большинства пациентов низкая, вплоть до нескольких лет.
Синдром лея что это такое прогноз
а) Терминология:
1. Сокращения:
• Синдром Лея (ЛС)
2. Синонимы:
• Подострая некротизирующая энцефаломиелопатия
3. Определение:
• Генетически гетерогенное митохондриальное заболевание, характеризующееся прогрессирующей нейродегенерацией
2. КТ признаки синдрома Лея:
• Бесконтрастная КТ:
о Низкая плотность; иногда нормальная рентгенологическая картина
• КТ с контрастированием
о Накопление контрастного вещества нехарактерно
3. МРТ признаки синдрома Лея:
• Т1-ВИ:
о Гипоинтенсивный сигнал:
— Возможно наличие гиперинтенсивных участков = кровь или некроз
• Т2-ВИ:
о Гиперинтенсивный сигнал
• FLAIR:
о Гиперинтенсивный сигнал:
— В хроническую стадию заболевания может наблюдаться разрешение участков изменения сигнала или кистозной энцефаломаляции (гипоинтенсивный сигнал)
• ДВИ:
о Ограничение диффузии в зоне острого поражения
• МР-спектроскопия:
о ↑ пика холина, ↓ пика NAA
о Часто присутствует пик лактата; может быть высоким
4. УЗИ:
• Гиперэхогенность глубоких ядер, БВ
5. Рекомендации по визуализации:
• Лучший инструмент визуализации:
о МРТ с ДВИ/МР-спектроскопия
в) Дифференциальная диагностика синдрома Лея:
2. Митохондриальная энцефалопатия, лактоацидоз, инсультоподобные эпизоды (MELAS):
• ↑ интенсивности сигнала на T2-BИ/FLAIR от скорлупы (кальцификация в хроническую стадию)
о Возможна асимметричность или односторонность поражения
• Инсультоподобные изменения сигнальных характеристик в теменно-затылочных областях полушарий:
о Характерно несоответствие бассейнам кровоснабжения и ДВИ(-)
3. Глутаровая ацидурия 1-го типа (ГА1):
• ↑ интенсивности сигнала на T2-BИ/FLAIR от полосатых тел, БШ ± поражение БВ
• Характерное расширение височных покрышек
4. Болезнь Вильсона-Коновалова:
• ↑ интенсивности сигнала на Т2-ВИ/FLAIR от скорлупы, БШ, средний мозг, таламусы:
о Изменения на Т2-ВИ очевидны у детей более старшего возраста, подростков
• Гиперинтенсивный на Т1-ВИ сигнал от БШ вследствие печеночной недостаточности
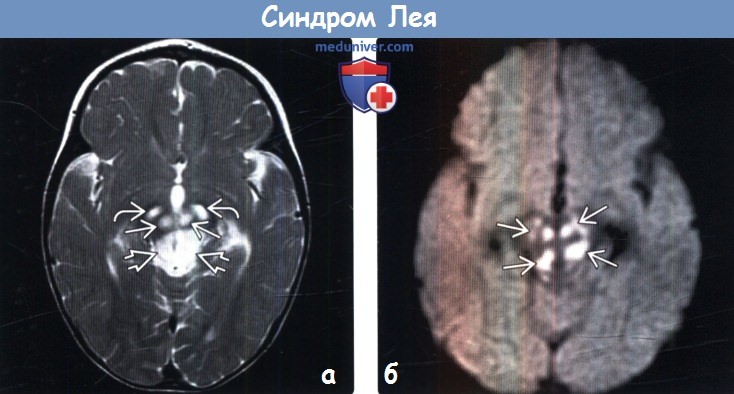 (а) МРТ, Т2-ВИ, аксиальный срез: отмечается повышение интенсивности сигнала от ножек мозга, красных ядер и покрышки среднего мозга (включая периакведуктальное серое вещество). Эти структуры являются частой локализацией поражения ствола мозга при синдроме Лея.
(а) МРТ, Т2-ВИ, аксиальный срез: отмечается повышение интенсивности сигнала от ножек мозга, красных ядер и покрышки среднего мозга (включая периакведуктальное серое вещество). Эти структуры являются частой локализацией поражения ствола мозга при синдроме Лея.
(б) МРТ, ДВИ, аксиальный срез: определяется ограничение диффузии (гиперинтенсивные участки) в пораженных участках среднего мозга. Ограничение диффузии указывает на острую стадию поражения, в то время как повышение диффузии больше указывает на хроническую стадию поражения.
2. Макроскопические и хирургические особенности:
• Коричневато-серые желатинозные или полостные участки в полосатых телах, БШ, СтМ, зубчатых ядрах, таламусах, спинном мозге, белом веществе
3. Микроскопия:
• Спонгиозная дегенерация, глиоз, потеря нейронов, демиелиниза-ция, пролиферация капилляров
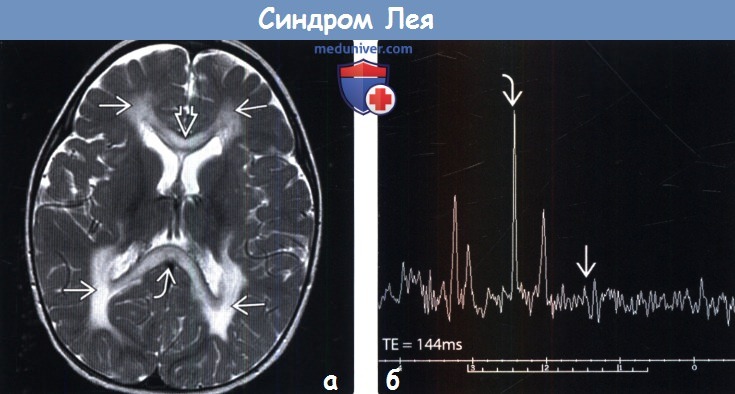 (а) МРТ, Т2-ВИ, аксиальный срез: определяется повышение интенсивности сигнала от колена и валика мозолистого тела, распространяющееся на пери вентрикулярное и глубокое белое вещество, а также задние бедра внутренних капсул.
(а) МРТ, Т2-ВИ, аксиальный срез: определяется повышение интенсивности сигнала от колена и валика мозолистого тела, распространяющееся на пери вентрикулярное и глубокое белое вещество, а также задние бедра внутренних капсул.
(б) Одновоксельная протонная МР-спектроскопия (ТЕ = 144 мс): у того же пациента определяется минимальный пик лактата на 1,33 ppm и аномальный пик на 2,4 ppm, что соответствует сукцинату. Был поставлен диагноз «дефицит сукцинатдегидрогеназы вследствие БСНА-мутации».
д) Клиническая картина:
1. Проявления синдрома Лея:
• Наиболее частые признаки/симптомы:
о Проявления: задержка/регрессия психомоторного развития, гипотония
— Синдром Лея (СЛ) является клиническим диагнозом; вызывать данные проявления могут многие митохондриальные заболевания
о Другие признаки/симптомы:
— Прогрессирующая дисфункция СтМ и БГ:
Атаксия, офтальмоплегия, птоз, рвота, нарушения проглатывания и дыхания, дистония
о Раннее проявление заболевания, дисфункция СтМ, периферическая нейропатия и быстрое неврологическое ухудшение характерно для СЛ, вызванного SURF1-мутацией
о Метаболические стрессовые факторы (например, инфекция) могут «демаскировать» заболевание или вызвать ее ухудшение о Повышение лактата в СМЖ, сыворотке крови, моче является классическим признаком, но наблюдается не всегда
о Клинический диагноз:
— Прогрессирующая нейродегенерация
— Признаки/симптомы дисфункции СтМ и БГ
— ↑ лактата в крови + СМЖ
— Биохимический дефект, выявленный с помощью митохондриального анализа биоптата скелетных мышц или культивируемых фибробластов кожи
— МРТ → характерные поражения БГ или СтМ
о Пренатальная диагностика: проба ворсинчатого хориона (мутации и биохимические дефекты)
• Клинический профиль:
о Младенец с психомоторной регрессией, гипотонией
2. Демография:
• Возраст:
о У большинства заболевание проявляется в возрасте двух лет
о Манифестация заболевания в более старшем детском и взрослом возрасте встречается нечасто
• Половая принадлежность:
о Отсутствует
• Этническая принадлежность:
о Отсутствует
• Эпидемиология:
о Митохондриальные заболевания = 1: 8500
о СЛ у детей
Редактор: Искандер Милевски. Дата публикации: 21.4.2019