Синдром крузона что это такое
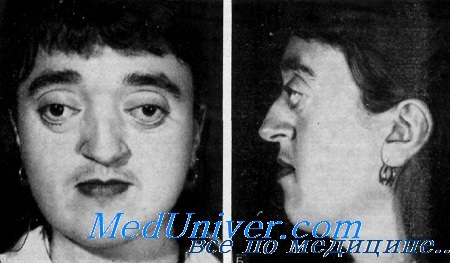
Черепно-лицевой дизостоз характеризуется преждевременным синостозированием черепа, гипоплазией средней части лица с относительным нижнечелюстным прогматизмом, экзофтальмом и гипертелоризмом.
Клинические данные. Данные осмотра. Клинические изменения ограничены черепом и лицом. Нос может иметь несколько необычную форму в связи с вертикальной гипоплазией средней трети лица. Часто наблюдаются относительный нижнечелюстной прогматизм, опущенная нижняя губа, короткая верхняя губа и торчащий язык. Постоянной чертой является неполное смыкание зубов.
Костная система. Форма черепа зависит от того, какие швы вовлечены в процесс. Могут наблюдаться брахицефалия, скафоцефалия, тригоноцефалия и изредка аномалия Kleeblattschadel. Обычно отчетливо пальпируются утолщенные края швов. Около брегмы (места соединения венечного и стрелковидного швов черепа) могут наблюдаться экзостозы (Bertelsen). Преждевременный краниосиностоз варьирует по времени начала, но чаще всего он развивается в течение первого года и заканчивается к 2—3 годам жизни. В некоторых случаях синостоза может не быть до 10-летнего возраста.
Орган зрения. Экзофтальм является вторичным. Он обусловлен уменьшением глубины орбит. Отмечаются расходящееся косоглазие и нистагм. Постоянной чертой является гипертелоризм. Bertelsen отметил у 80% больных с черепно-лицевым дизостозом поражение зрительного нерва. Иногда могут также наблюдаться спонтанный вывих глазного яблока, мегалокорнеа, эктопия хрусталика, колобома радужной оболочки и эктопия зрачка.
Орган слуха. Boedts установил, что у 1/3 больных с синдромом Крузона имеется глухота, чаще проводящего типа. При хирургическом и посмертном исследовании он обнаружил деформацию слуховых косточек и фиксацию стремени в овальном окне. Aubrey, Nager и de Reynier, Wiegand и Baldwin отметили двустороннюю атрезию наружного слухового прохода и смешанную или проводящую глухоту. Schurmans и Hariga выявили снижение костной проводимости, отсутствие восстановления и посредством томографии деформацию внутреннего слухового прохода.
Вестибулярная система. Aubrey описал нормальную вестибулярную функцию.
Лабораторные данные. Рентгенограммы. Чаще всего преждевременным синостозированием охвачены коронарный, сагиттальный и лямбдовидный швы. Кроме этого, могут наблюдаться и другие рентгенологические находки, такие, как пальцевые вдавления, уплощение глазниц, базилярный кифоз, расширение ямки гипофиза и маленькие параназальные синусы (Bertelsen). Как отметили Schurmans и Hariga, при томографическом исследовании наблюдается деформация внутреннего слухового прохода. Рентгенография показала нормально развитые лабиринты (Wiegand). Изящное томографическое исследование височной кости, проведенное Terrahe, обнаружило наружную ротацию каменистой части пирамиды, вторичную по отношению к дисплазии основания черепа.
В результате этого слуховые каналы имели косое направление, неправильным было и направление лицевого нерва, кроме того, наблюдались гиперостозы. Terrahe подчеркнул, что первичными изменениями являлись фиксация слуховых косточек со скоплением костных масс внутри барабанной полости, аномалии слуховых косточек и закрытие овального окна.
Патология. Томография височной кости обнаружила стеноз или атрезию наружного слухового прохода, отсутствие барабанной полости, деформацию стремени и костное синостозирование его с мысом, анкилоз молоточка с наружной стенкой верхней части барабанной полости, искривление и сужение среднего уха, а также воздухоносных пространств сосцевидного отростка (Nager, de Reynier). Baldwin обнаружил также недоразвитие периостальной части лабиринта. Молоточек и наковальня были анкилознрованы к боковой стенке пиши эпитимпанума. Ножки стремени были косо направлены к основанию, сустав между стременем и наковальней соприкасался с мысом. И круглое, и овальное окна были сужены. Барабанная перепонка отсутствовала.
Наследственность. Наследование аутосомно-доминантное с полной пенетрантностью (Schiller, Vulliamy, Normandale). Почти 1/3 случаев — новые мутации.
Диагноз. Черепно-лицевой дизостоз должен быть отграничен от изолированного крапиостеноза, синдрома Апера, синдрома Пфейффера и синдрома Сетре—Чотцена (Gorlin et al.).
Лечение. Могут быть произведены косметическая и функциональная коррекция посредством удаления по частям преждевременно синостозированных швов и хирургическое создание блефарофимоза с целью предотвращения вывиха глазного яблока. Tessier описал превосходную радикальную хирургическую операцию для исправления деформаций лица.
Прогноз. С возрастом часто становится невозможным бинокулярное зрение и отмечается расходящееся, альтернирующее и содружественное косоглазие. Нередко наблюдается атрофия зрительного нерва и потеря зрения. Иногда встречается полное выпадение одного или обоих глазных яблок, происходящее вследствие уплощения глазниц. Преждевременное синостозирование швов ведет к образованию экзостозов в области брегмы. С возрастом гипоплазия средней части лица может становиться все более выраженной при нормальном росте нижней челюсти.
Выводы. Характеристика синдрома Крузона включает:
1) аутосомно-доминантное наследование;
2) преждевременный варьирующий краниосиностоз;
3) гппертелоризм, уплощение глазниц и экзофтальм;
4) клювовидный нос;
5) гипоплазию верхней челюсти с относительным нижнечелюстным прогнатизмом;
6) иногда двустороннюю атрезию наружных слуховых проходов, аномалии слуховых косточек и смешанную глухоту.
Лечение синдрома Крузона в Израиле с применением современных консервативных и хирургических методов
Синдром Крузона представляет собой крайне редкую врожденную патологию, причины которой заключаются в формировании генных мутаций. Характерные признаки заболевания выражаются в краниосиностозе, или преждевременном сращении костей черепа, в результате чего деформируются лицевой и мозговой отделы. Поскольку улучшить в целом неблагоприятный прогноз данного синдрома возможно только при своевременном проведении адекватной терапии, ее разработка требует высочайшего уровня квалификации и большого опыта специалистов. Лечение синдрома Крузона в Израиле проходит согласно комплексной программе, в составлении и осуществлении которой принимает участие группа узкопрофильных врачей.
 Современное лечебно-диагностическое оборудование диагностических центров Израиля дает возможность проведения буквально за несколько дней всестороннего обследования пациента. На основе полученных показателей специалисты подбирают наиболее эффективные для данного больного современные методы терапии и лекарственные препараты последнего поколения. Нейрохирурги и пластические хирурги мирового уровня квалификации выполняют малотравматичные операции, направленные на ликвидацию образовавшихся дефектов и развившихся осложнений. Отзывы больных и их родителей подтверждают, что, даже при условии неизлечимости болезни, назначаемый лечебный курс успешно купирует симптомы и создает условия для полноценного развития ребенка. При этом подчеркивается приемлемая стоимость медицинских услуг.
Современное лечебно-диагностическое оборудование диагностических центров Израиля дает возможность проведения буквально за несколько дней всестороннего обследования пациента. На основе полученных показателей специалисты подбирают наиболее эффективные для данного больного современные методы терапии и лекарственные препараты последнего поколения. Нейрохирурги и пластические хирурги мирового уровня квалификации выполняют малотравматичные операции, направленные на ликвидацию образовавшихся дефектов и развившихся осложнений. Отзывы больных и их родителей подтверждают, что, даже при условии неизлечимости болезни, назначаемый лечебный курс успешно купирует симптомы и создает условия для полноценного развития ребенка. При этом подчеркивается приемлемая стоимость медицинских услуг.
Методы лечения патологии
Синдром Крузона был описан более ста лет назад, но проведенные за это время глубокие исследования проблемы так и не позволили окончательно выяснить причины возникновения лежащих в его основе мутаций. Выяснилось, что достаточно часто заболевание развивается в результате спонтанно возникающих мутационных изменений гена, отвечающего за формирование костной ткани и окостенение.
Подозрения на наличие синдрома Крузона часто возникают у врачей при осмотре новорожденного ребенка, однако отчетливо признаки патологии проявляются на протяжении первых лет жизни малыша. Развивающийся краниосиностоз вызывает замедление, а при дальнейшем прогрессировании, полную остановку нормального роста и развития головы. После рождения у ребенка заметны гипертелоризм, или чрезмерное расстояние между глазницами, пучеглазие (экзофтальм), патологический прикус (прогнатия нижней челюсти), заостренный нос. У некоторых пациентов выявляется атрезия хоан — заращение носовой полости костной и хрящевой тканью, делающее невозможным дыхание носом.
Прогрессирование патологического процесса приводит к развитию неврологического синдрома и отставания в интеллектуальном развитии. В результате повышения внутричерепного давления у больного возникают гидроцефалия, интенсивные головные боли, тошнота с приступами рвоты, приступы судорог.
Несмотря на бурное развитие медицины и фармакологии, схемы специфического лечения синдрома Крузона не разработаны. Такт как патология является неизлечимой, показаны паллиативные терапевтические меры, целью которых является купирование симптоматики и устранение образовавшихся дефектов.
Медикаментозное лечение
В зависимости от выраженных симптомов в лечебную схему включаются следующие виды медикаментозных препаратов:
Хирургическое лечение
Хирургические операции проводятся с целью устранения последствий черепного синостоза и коррекцию патологических изменений формы черепа. Для максимальной эффективности вмешательство проводится на протяжении первого года жизни ребенка, а в дальнейшем повторно выполняются до момента завершения роста черепных костей. При своевременном хирургическом лечении в большинстве случаев удается избежать развития неврологического синдрома, внутричерепной гипертензии, отставания в умственном развитии.
Для корректировки деформации костей черепа выполняется краниопластика. В ходе этой оперативной процедуры иссекаются костные швы черепной коробки, что приводит к увеличению ее объема и улучшению формы черепа. В случае выраженного экзофтальма хирургическим путем корректируется глубина глазных орбит и промежуток между ними. Имплантирование шунта в мозговой желудочек для оттока накапливающихся излишков спинномозговой жидкости способствует стабилизации нормальных показателей внутричерепного давления и устранению гидроцефалии. Корректировка смещения мозжечка, выявляющегося у пациентов с данным синдромом, проводится путем увеличения размеров черепной ямки, где локализован орган.
В случае крайнего затруднения дыхательных движений и прохождения воздуха при атрезии хоан у новорожденных и грудных малышей выполняется трахеостомия. В старшем детском возрасте рекомендуется хирургическое исправление дефектов лицевого отдела, воссоздание правильного прикуса.
Способы диагностики патологии
Выявление синдрома Крузона возможно при проведении пренатальной генетической диагностики. Для диагностирования патологии у новорожденного младенца или ребенка младшего возраста необходимо проведение комплексного обследования, включающего инструментальные и лабораторные методы. В Израиле всестороннее обследование пациента, постановка диагноза и выстраивание схемы лечения занимает около трех дней.
На первичной консультации ведущего неонатолога или педиатра, на которую пациента направляют сразу после поступления в клинику, врач знакомится с историей болезни, результатами более ранних исследований и медицинскими заключениями, расспрашивает родителей ребенка о характере проявляющихся симптомов. При поверхностном осмотре выявляются характерные внешние дефекты. В обязательном порядке проводится офтальмологический осмотр, проверка слуха, оценивается уровень умственного развития. На приеме врач назначает требующиеся обследования.
Выполнение указанных в списке назначений обследований:
— молекулярно-генетическое тестирование — наиболее показательная диагностическая методика, целью которой является выявление определенной генной мутации;
— рентгенографическое исследование черепа;
— компьютерная томография (КТ) черепа.
Результаты исследований передаются на рассмотрение врачебного консилиума, состоящего из неонатолога, педиатра и узкопрофильных специалистов. После анализа данных врачи коллегиально устанавливают диагноз и разрабатывают терапевтическую программу.
Сколько стоит лечение патологии
Учитывая сложность лечения, его цена волнует большинство медицинских туристов. Прохождение терапии в израильских медцентрах обеспечивает экономию около 30% суммы, необходимой в странах Западной Европы, и стоит примерно на 50% дешевле аналогичных услуг в США.
Преимущества лечения в Израиле
При условии своевременного прохождения полноценной терапии состояние маленького пациента заметно улучшается, и он обретает возможность нормального развития. Обратившись в израильскую клинику, вы доверите здоровье малыша высококвалифицированным специалистам, обладающим всеми материально-техническими возможностями для качественного лечения.
Синдром Крузона
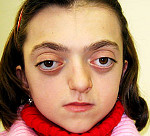
Синдром Крузона – редкое генетическое заболевание, сопровождающееся прогрессирующими деформациями лицевой и мозговой части черепа и краниосиностозом с развитием сопутствующих нарушений. Симптомами этого состояния являются изменение формы головы (брахицефалия, скафоцефалия, тригоноцефалия), крючковидный нос, гипоплазия средней трети лица, нарушения зрения и слуха. Диагностика синдрома Крузона осуществляется на основании внешних проявлений заболевания, рентгенологических данных, а также молекулярно-генетических анализов. Специфического лечения этой патологии не существует, используются паллиативные и симптоматические мероприятия, в том числе хирургического характера.
Общие сведения
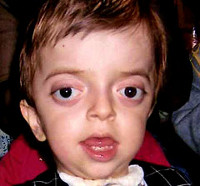
Синдром Крузона (краниофасциальный дизостоз 1 типа) – генетическое заболевание, характеризующееся нарушением процессов окостенения и развития элементов скелета лицевого и мозгового черепа. Впервые это состояние было описано в 1912 году французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона – аутосомно-доминантный, однако заболевание часто обусловлено спонтанными мутациями. Патология встречается достаточно редко – примерно 1,6 случаев на 100 000 новорожденных, при этом данным синдромом обусловлено почти 5% от всех пороков развития, сопровождающихся черепным дизостозом. Долгое время считалось, что это состояние имеет две разновидности – обычную и сопровождающуюся кожными нарушениями (гиперкератозом, акантозом), но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (CAN) является отдельным наследственным заболеванием. В то же время, патогенез его развития аналогичен классической форме заболевания, именно этим объясняется значительная схожесть симптомов. Состояние с одинаковой вероятностью поражает как мальчиков, так и девочек.
Причины синдрома Крузона
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме – он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность – в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета. Так, помимо синдрома Крузона, мутации FGFR2 могут быть причиной синдромов Апера, Сетре-Чотзена, Бира-Стивенсона, синдрома Пфайффера и многих других патологий. Генетические исследования показали, что краниофасциальный дизостоз 1-го типа способны вызывать более 35 мутаций вышеуказанного гена, в основном они локализованы в области 7 и 9 экзонов.
Практически все дефекты гена FGFR2 относятся к миссенс-мутациям, то есть провоцируют изменение структуры кодируемого белка. Изменение конформации рецептора к фактору роста фибробластов 2 нарушает межклеточные взаимодействия в соединительных тканях черепа, главным образом костной и хрящевой. Это приводит сначала к накоплению фибробластов в области межкостных швов, а потом к активизации процессов окостенения, что и является причиной ведущего проявления синдрома Крузона – черепного синостоза. Некоторые исследователи полагают, что данные генетические дефекты влияют также на эмбриональное развитие структур первой жаберной дуги – к ним относят челюсти и отчасти элементы средней трети лица. Именно этим объясняется гипоплазия челюстей, особенно нижней, при синдроме Крузона.
Причины синдрома Крузона с черным акантозом несколько иные – он вызывается мутациями гена FGFR3, локализованного на 4 хромосоме. Продуктом его экспрессии также является рецептор к фактору роста фибробластов, только 3 типа (в отличие от 2 типа, являющего продуктом гена FGFR2). Выяснено, что только одна мутация этого гена выступает причиной синдрома Крузона с характерными кожными проявлениями – Ala391Glu. Это тоже миссенс-мутация, изменяющая структуру белка-рецептора. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом аналогичны предыдущему типу, однако к ним присоединяются гиперкератоз различных участков кожи и акантоз, нередко наблюдаются многочисленные родинки.
Симптомы синдрома Крузона
Проявления синдрома Крузона можно заметить уже при рождении ребенка, однако наиболее выраженными они становятся на протяжении первых 3-4 лет жизни. Самым характерным симптомом заболевания является краниосиностоз, который может развиваться на венечном или стреловидном (намного реже) шве, прочно соединяя кости и останавливая нормальный рост головы. Сразу после рождения первые признаки синостоза могут быть стертыми, но всегда наблюдается гипертелоризм, прогнатия нижней челюсти, изменение формы носа по типу «клюва попугая», незначительный экзофтальм из-за уменьшенного размера глазниц, низкое расположение наружного слухового прохода. Иногда при синдроме Крузона выявляется синдактилия пальцев, в этом случае необходимо производить дифференциальную диагностику с синдромом Апера. У некоторых больных обнаруживается атрезия хоан, затрудняющая дыхание, а также гидроцефалия, еще больше осложняющая течение заболевания за счет резкого возрастания внутричерепного давления.
Особенностью синдрома Крузона является неминуемое прогрессирование заболевания, особенно в отношении формы черепа. Из-за образования прочного синостоза и продолжающегося роста размеров головного мозга форма головы изменяется, возникает брахицефалия или «башенный череп» – в зависимости от того, по какому шву произошло срастание. При синдроме Крузона в области сросшихся костей черепа также могут образовываться экзостозы. Такая деформация приводит и к поражению органов зрения – сначала возникает расходящееся косоглазие, затем экзофтальм сильно прогрессирует вплоть до выпадения глазных яблок из орбиты. Нередко синдром Крузона сопровождается расстройствами слуха из-за нарушения структуры пирамиды височной кости – ее полости уменьшены в размерах, некоторые из них могут отсутствовать, нередко это приводит к полной глухоте. Наблюдаются изменения и со стороны нервной системы, обнаруживаются нарастающие признаки умственной отсталости (при отсутствии паллиативных мероприятий), симптомы повышения внутричерепного давления (головные боли, рвота), судорожные припадки.
Синдром Крузона с черным акантозом характеризуется аналогичными изменениями со стороны лица и черепа. При этом некоторые исследователи отмечают, что данная форма заболевания протекает в целом тяжелее и характеризуется повышенной частотой осложнений. Так, атрезия хоан, довольно редко развивающаяся при классической разновидности краниофасциального дизостоза 1 типа, в случае синдрома Крузона с черным акантозом регистрируется почти у половины больных. Кроме того, у пациентов наблюдаются сильно выраженные кожные нарушения – гиперкератоз (разрастание бородавок, гипертрофия кожи), усиленная пигментация. Основная локализация кожных проявлений при синдроме Крузона с черным акантозом – области коленных и локтевых сгибов, шея, живот, носогубные складки, зона вокруг глаз. Также для этого заболевании характерно наличие большого количества невусов (родинок), часто развиваются гипертрофические слабо пигментированные рубцы и шрамы.
Диагностика синдрома Крузона
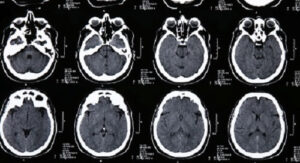
Выявление синдрома Крузона возможно на этапе пренатального развития, сразу после рождения или в первые годы жизни больного. Для этого применяются рентгенологические методики, общий осмотр, молекулярно-генетические анализы. Вспомогательную роль в диагностике синдрома Крузона играют такие методы, как офтальмологический осмотр, исследование слуха, оценка интеллектуального и психического развития. При осмотре маленьких детей определяются низко посаженные уши, гипоплазия средней трети лица, экзофтальм. У больных синдромом Крузона старшего возраста к этим проявлениям присоединяются расходящееся косоглазие, ослабление слуха вплоть до полной глухоты, изменение формы черепа. На рентгенографии черепа регистрируется синостоз в области венечного, стреловидного или лямбдовидного швов, возможно обнаружение экзостозов и уплощенной формы глазниц.
Томография пирамиды височной кости при синдроме Крузона выявляет нарушение формирования наружного слухового прохода (атрезия или стеноз) и других полостей, иногда наблюдается отсутствие барабанной полости. Турецкое седло несколько расширено, могут образовываться добавочные мелкие околоносовые синусы. Молекулярно-генетическая диагностика синдрома Крузона производится врачом-генетиком и при классической форме заболевания сводится к автоматическому секвенированию 7 и 9 экзонов гена FGFR2 с целью выявления мутаций. При наличии кожных проявлений (гиперкератоза, бородавках, множественных родинках) имеет смысл производить поиск мутации Ala391Glu в гене FGFR3. Для обеих форм синдрома Крузона возможна пренатальная генетическая диагностика, ультразвуковые методики при этом, как правило, малоэффективны.
Лечение синдрома Крузона
Какого-либо специфического лечения синдрома Крузона на сегодняшний момент не существует, применяют только паллиативные мероприятия. К ним относят хирургические вмешательства по ремоделированию формы черепа и устранению синостозов – такие процедуры необходимо начинать как можно раньше и в дальнейшем производить еще несколько раз по мере роста головы. Это снижает уровень внутричерепного давления, что положительно сказывается на умственном развитии больных синдромом Крузона и уменьшает вероятность появления неврологических нарушений. Также с помощью хирургических методик создают искусственный блефарофимоз для снижения степени экзофтальма и предотвращения вывиха глазного яблока. При атрезии хоан производится их расширение оперативным путем для облегчения дыхания. Описаны техники радикальных комплексных операций, направленных на устранение большинства лицевых нарушений при синдроме Крузона. В случае развития кожных изменений для снижения их выраженности рекомендуется наружное применение средств на основе ретиноидов, иногда назначают кортикостероиды.
Прогноз и профилактика синдрома Крузона
Прогноз синдрома Крузона, как правило, неопределенный, многие специалисты оценивают его как неблагоприятный. Это связано с тем, что даже при проведении всех симптоматических и паллиативных мероприятий у больных все равно нарастает расходящееся косоглазие, практически всегда со временем развивается глухота, гипоплазия средней трети лица становится более выраженной с возрастом. Тем не менее, многие больные при соответствующем лечении и уходе могут доживать до преклонного возраста. По причине сильного нарушения зрения и слуха практически всегда происходит инвалидизация пациентов, причиной инвалидности также может стать умственная отсталость. Профилактика синдрома Крузона не разработана, возможно лишь пренатальное определение патологии молекулярно-генетическими методами.
Синдром Крузона

Ведущий специалист отделения хирургии молочной железы
онлайн-консультаций с израильским экспертом организовываются в течение 2-3 дней
пациентов выбирают онлайн-консультацию израильского эксперта еще до приезда в клинику
пациентов выбирают онлайн-поддержку от медицинских консультантов нашей клиники еще до приезда в Израиль
 >Синдром Крузона является диагностирующимся у малого количества пациентов наследственным заболеванием. Клинические признаки патологии заключаются, прежде всего, в краниосиностозе — процессе раннего сращения черепных костей, приводящем к выраженной деформации мозговой и лицевой части черепа. При отсутствии адекватной комплексной терапии большинство специалистов отмечают неблагоприятный прогноз данной патологии. Как и в случае других генетических заболеваний, замедление прогрессирования нарушений требует высокой квалификации врачей и проведения процедур на высокотехнологичной аппаратуре. Лечение синдрома Крузона в Израиле проводится в соответствии с этими требованиями, по комплексной схеме, индивидуально разрабатываемой для каждого пациента.
>Синдром Крузона является диагностирующимся у малого количества пациентов наследственным заболеванием. Клинические признаки патологии заключаются, прежде всего, в краниосиностозе — процессе раннего сращения черепных костей, приводящем к выраженной деформации мозговой и лицевой части черепа. При отсутствии адекватной комплексной терапии большинство специалистов отмечают неблагоприятный прогноз данной патологии. Как и в случае других генетических заболеваний, замедление прогрессирования нарушений требует высокой квалификации врачей и проведения процедур на высокотехнологичной аппаратуре. Лечение синдрома Крузона в Израиле проводится в соответствии с этими требованиями, по комплексной схеме, индивидуально разрабатываемой для каждого пациента.
Оснащение израильских медицинских центров высокоточной новейшей аппаратурой позволяет оперативно выполнить диагностическое обследование, требующееся для постановки правильного диагноза. Большое значение для разработки действенного лечения имеет мультидисциплинарный подход, предполагающий разработку терапевтической схемы и подбор включаемых в нее методик комиссией, в которую входит ряд узкопрофильных специалистов. Несмотря на неизлечимость заболевания, отзывы пациентов свидетельствуют о том, что проводимая в Израиле терапия купирует болезненные симптомы, нормализует общее состояние и позволяет ребенку полноценно развиваться. Больным обеспечивается максимальный комфорт, с ними работает группа психологов. При этом отмечается лояльная стоимость лечения, которая вполне соответствует качеству оказываемых услуг.
Методы лечения заболевания
Синдром Крузона развивается в результате возникновения определенной генной мутации. Патология была впервые описана в начале прошлого столетия, однако проведенные за минувшие годы многочисленные исследования так и не указали специалистам на точные причины этой мутации. Этот диагноз ставится крайне редко, примерно у 1-2 на 100 000 новорожденных детей.
В результате краниосиностоза замедляется, а постепенно практически останавливается, рост и нормальное развитие головы. К характерным признакам относится гипертелоризм (расположение глазниц на слишком большом расстоянии друг от друга), экзофтальм (пучеглазие), выступание вперед верхней челюсти и ее нависание над нижней (прогнатия), остроконечный нос, похожий на клюв попугая, плоские виски и лоб при условии маленького вытянутого лица. Дефекты костей черепа прогрессируют, сопровождаясь развитием неврологических осложнений и задержки умственного развития. Чрезмерные показатели внутричерепного давления вызывают у пациента сильные головные боли и рвоту, часто наблюдаются судорожные припадки, гидроцефалия. Кроме того, у многих больных развивается сужение носовых проходов, затрудняющее, а подчас делающее невозможным, дыхание через нос.
На сегодняшний день не существует специфической терапии патологии. Поскольку синдром Крузона входит в число неизлечимых заболеваний, показаны паллиативные лечебные процедуры, направленные на устранение развившихся дефектов и нормализацию состояния пациента.
Медикаментозная терапия
Различные виды действенных лекарственных средств подбираются в зависимости от проявляющейся симптоматики болезни:
Хирургическое лечение
Оперативные вмешательства направлены на устранение синостозов и коррекцию измененной формы черепа. Для достижения положительного результата операции должны проводиться уже в первый год жизни пациента, после чего повторно выполняться до завершения роста костей черепа. Таким образом удается успешно избежать повышения внутричерепного давления, развития неврологических нарушений, умственной отсталости.
Деформация костей черепной коробки корректируется с помощью краниопластики. В ходе этого оперативного вмешательства проводится иссечение швов между черепными костями, в результате чего увеличивается объем черепной коробки и исправляется форма черепа. При тяжелой степени экзофтальма проводится хирургическая коррекция глубины орбит глаза и расстояния между ними. Для устранения гидроцефалии и нормализации внутричерепного давления в желудочек головного мозга имплантируется шунт, предназначенный для отведения чрезмерного количества ликвора. Развивающееся при синдроме Крузона смещение мозжечка корректируется с помощью хирургического увеличения черепной ямки, в которой располагаются его структуры.
Большое внимание уделяется нормализации дыхания, крайне затрудненного в результате сужения носовых проходов. В случае тяжелых нарушениях дыхательной функции у новорожденных и детей в возрасте до года показана трахеостомия. Пациентам старшего детского возраста выполняется хирургическая коррекция лицевого отдела черепа. При этом устраняются дефекты положения челюстей и других лицевых структур.
Как диагностируется заболевание
Правильный диагноз возможно поставить в результате комплексного обследования пациента, на выполнение которого в израильских клиниках требуется, в среднем, три дня.
В первый день пребывания в клинике пациент должен посетить лечащего врача. В ходе первичной консультации специалист изучает историю болезни и предоставленную медицинскую документацию (результаты проводимых на родине больного исследований, врачебные заключения), проводит тщательный осмотр. Осмотр маленьких пациентов позволяет выявить характерные для данной патологии внешние дефекты. Кроме того, необходим офтальмологический осмотр, проверка остроты слуха, оценка уровня интеллектуального развития. Прием завершается назначением требующихся обследований.
Следующий день посвящен выполнению назначенных лечащим врачом обследований:
После получения результатов обследований их изучает врачебный консилиум, в состав которого входят лечащий врач и узкопрофильные специалисты. Врачи определяются с диагнозом и разрабатывают индивидуальную схему лечения.
Сколько стоит лечение заболевания
Цена терапии, итоговый размер которой определяется только после проведения обследования и назначения лечебных процедур, интересует практически всех медицинских туристов. Как отмечают, лечение зарубежных пациентов в Израиле позволяет им сэкономить примерно 30% от суммы, требуемой в западноевропейских клиниках, и около 50% необходимой в США.
Преимущества лечения в Израиле
Несмотря на то, что синдром Крузона относится к неизлечимым заболеваниям, своевременное проведение адекватной терапии позволяет пациенту нормально развиваться и вести полноценную жизнь. В израильских клиниках существуют все условия для эффективного лечения, поэтому связывайтесь с выбранным медцентром и восстанавливайте здоровье под руководством компетентных врачей.