Синдром Криглера – Найяра
Синдром Криглера – Найяра – наследственная гипербилирубинемия (повышение уровня непрямого билирубина в крови), для которой характерна желтуха и тяжелое поражение нервной системы. Развитие данной патологии связано с отсутствием в клетках печени фермента уридиндифосфат-глюкуронилтрансферазы (УДФГТ). Из-за этого орган не способен конъюгировать билирубин, т. е. преобразовывать свободный билирубин в малотоксичный прямой. Выделяют два типа данного синдрома, различающиеся тяжестью симптомов.
Причины возникновения
Развитие синдрома Криглера – Найяра обусловлено генетическими факторами. Патология возникает при мутациях гена, который кодирует фермент печени УДФГТ. При синдроме I типа уридиндифосфат-глюкуронилтрансфераза отсутствует полностью, что приводит к появлению тяжелой симптоматики сразу после рождения ребенка. В результате критического повышения уровня билирубина развивается поражение головного мозга, что ведет к инвалидизации. При синдроме II типа генетические дефекты приводят к снижению активности УДФГТ печени. Тем не менее этот фермент присутствует, поэтому патология сопровождается менее выраженными проявлениями.
Симптомы
При синдроме II типа. Патология может обнаружиться в первые месяцы жизни, а у некоторых больных она никак не проявляет себя до подросткового возраста. Иногда клиническая симптоматика полностью отсутствует. В целом симптомы могут быть такими же, как и при I типе, но менее выраженными. Неврологические осложнения встречаются редко. Билирубиновая энцефалопатия может развиться под влиянием стресса или инфекционных заболеваний.
Диагностика
Лабораторные методы диагностики. Чтобы исключить гемолитическую анемию, может быть назначен общий анализ крови. Важную роль в диагностике играет биохимическое исследование крови, позволяющее определить содержание непрямого билирубина в крови (при синдроме I типа – от 20 до 50 мг/дл, II типа – от 6 до 20 мг/дл). Дополнительно может назначаться проба с фенобарбиталом для выявления типа патологии.
Инструментальные методы диагностики. В рамках обследования для дифференциации (различения) типов синдрома может проводиться дуоденальное зондирование (получение выделившейся в ответ на введение раздражителя желчи при помощи зонда), хроматографический анализ желчи, пероральная холеграфия (рентген желчного пузыря), чрескожная биопсия печени.
Лечение
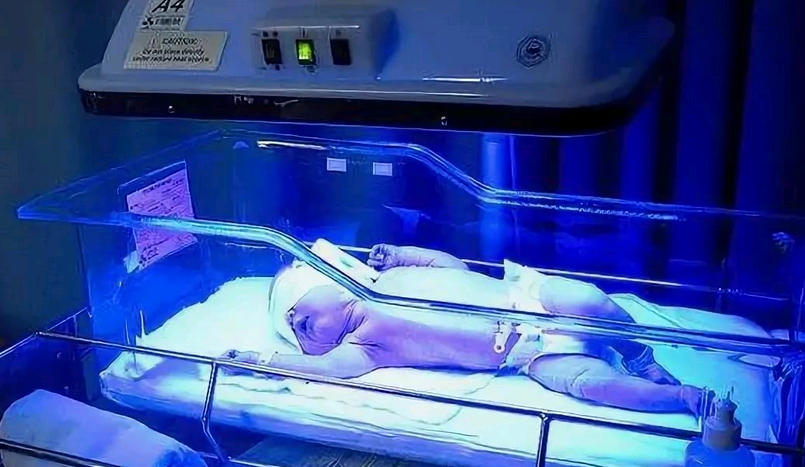 Фототерапия. Лечение сводится к облучению больного высокоинтенсивным светом с определенной длиной волны. Фототерапия призвана снизить токсическое воздействие билирубина и ускорить его выведение из организма с мочой.
Фототерапия. Лечение сводится к облучению больного высокоинтенсивным светом с определенной длиной волны. Фототерапия призвана снизить токсическое воздействие билирубина и ускорить его выведение из организма с мочой.
Введение плазмы крови и переливание крови. Введение плазмы может способствовать разрушению комплексов непрямого билирубина с альбумином и выведению билирубина (элиминации). Метод обычно сочетают с фототерапией. При неэффективности такого лечения может быть назначено переливание крови.
Плазмаферез. Удаление части плазмы крови может быть показано при синдроме II типа в период обострения. Плазмаферез может способствовать быстрому снижению концентрации непрямого билирубина.
Трансплантация печени. Данный метод направлен на нормализацию обмена билирубина в организме и может помочь значительно улучшить прогноз для больного.
Синдром криглера найяра что это
Синдром Жильбера— Леребулле, семейная гипербилирубинемия, связан с нарушением функции ферментов, обеспечивающих перенос непрямого билирубина из крови в печеночную клетку. Очевидно, происходит в какой-то степени и снижение процессов конъюнгации билирубина. Врожденное заболевание с аутосомно-доминантным типом наследования. Чаще болеют мальчики или взрослые мужчины, выявление болезни в периоде новорожденности маловероятно. Морфологические изменения в печени либо отсутствуют (у 0,25 больных), либо носят неспецифический характер.
Находят отложение липофусцина или жировую дистрофию гепатоцитов. Звездчатые эндотелиоциты образуют небольшие скопления.
При электронно-микроскопическом исследовании обнаруживается уменьшение числа и деформация микроворсинок на синусоидальном полюсе гепатоцитов.
Синдром Криглера — Найяра — тяжелое заболевание, связанное с врожденным отсутствием ферментов, участвующих в конъюгации билирубина. Наследуется аутосомно-рецессивно, встречается редко. Выявляется заболевание в периоде иоворожденности тяжелой желтухой, связанной с высоким (684 мкмоль/л — 40 мг% и более) содержанием непрямого билирубина.
Некоторые данные и, в частности, наличие обоих заболеваний в одних и тех же семьях свидетельствуют о близости синдромов Жильбера — Леребулле и Криглера — Найяра. Последний, по-видимому, является крайним вариантом первого.
Печень несколько увеличена лишь в редких случаях. Микроскопически выявляются желчные «тромбы» в канальцах, у части больных небольшой перипортальный фиброз или значительная жировая дистрофия гепатоцитов. При электронно-микроскопическом исследовании обнаруживается повреждение мембраны гепатоцита на синусоидальном полюсе с уменьшением числа микроворсинок и выходом органелл в перикатшллярные пространства (Диссе). Имеется также гиперплазия цитоплазматической сети, отложение желчного пигмента в гепатоцитах и эндотелиоцитах. Липофусцин в гепатоцитах не содержится.
Изменения мозга заключаются в ядерной желтухе с окрашивании гиппокамповой извилины, ядер ствола мозга и мозжечка. Имеются также очаги некроза в миокарде и скелетных мышцах, эрозии слизистой оболочки желудка и кишечника.
Преходящая семейная желтуха Люцея — Ариаса—Дрисколла, обменные или лекарственные гипербилирубинемии, проявляются в периоде иоворожденности (иногда на 7—9-е сутки) интенсивной желтухой с наличием в крови непрямого билирубина.
Нарушение конъюгации билирубина происходит при синдроме Люцея — Ариаса — Дрисколла в результате того, что на ферментные системы связывания билирубина Действует ингибитор, проникающий через плаценту от беременной женщины и являющийся, очевидно, прогестероном.
Аналогичное действие вызывают стероиды, в частности 3-а-20-бета-прегнандиол вырабатываемые в организме некоторых родильниц и поступающие в организм новорожденного с молоком. Прекращение кормления ребенка молоком матери ведет в этих случаях к быстрой нормализации билирубина, но возможно и тяжелое течение с развитием билирубиновой энцефалопатии. Как и при других желтухах, связанных с нарушением конъюгации билирубина, при синдроме Люцея — Ариаса — Дрисколла отсутствуют признаки гемолиза и грубые поражения печели. Отсутствует и плейохромия (интенсивная окраска) желчи и кала.
Ранняя желтуха новорожденного может быть также обусловлена поступлением через плаценту во время родов из организма беременной женщины эстрогенов, метаболитов, тормонов надпочечников и др. Связывание этих веществ происходит так же, как и непрямого билирубина. Перегрузка ферментных систем приводит к задержке выделения билирубина.
После рождения аналогичное конкурентное действие могут оказать многие лекарственные вещества (барбитураты, салицилаты, сульфаниламиды и др.).
Для конъюгации билирубина необходима глюкоза (в виде уридиндифосфатглюкозы), поэтому состояния, сопровождающиеся дефицитом глюкозы в организме новорожденного и гипогликемией, могут осложниться задержкой непрямого билирубина.
Криглера-Найяра синдром
OMIM 218800
Наша команда профессионалов ответит на ваши вопросы
Синдром Криглера-Найяра I типа (СК-Н I) – самая тяжелая форма заболевания из группы наследственных неконьюгированных гипербилирубинемий, обусловлен мутациями в кодирующей последовательности гена UGT1A1, что приводит к образованию неполноценного фермента уридиндифосфатглюкуронидазы 1, который разрушается. Для СК-Н I типа характерно полное отсутствие фермента УДФ-ГТ1 в гепатоцитах, в связи с чем реакции глюкуронизации билирубина не происходит и непрямой билирубин накапливается в организме, в том числе в ядрах серого вещества, обуславливая тяжелую клинику заболевания. В связи с отсутствием УДФ-ГТ1, фенобарбитал не имеет точки приложения и лечение данным препаратом неэффективно.
Данное заболевание характеризуется злокачественным прогрессирующим течением. Манифестация наступает в первые часы жизни. Клинические проявления заболевания: желтушность склер и кожных покровов, судороги, опистотонус, нистагм, атетоз, замедление умственного развития (билирубиновая энцефалопатия), на ЭЭГ регистрируется медленная активность в задних долях и параксизмальная активность. Биохимические показатели: уровень билирубина в крови выше 200 мкмоль/л. Гистологических изменений в печени не обнаруживают. В желчи полностью отсутствует коньюгированный билирубин. В отсутствии лечебных мероприятий больные погибают в течение первого года жизни от ядерной желтухи.
Синдром Криглера Найяра II (СК-Н II) типа занимает промежуточное положение по тяжести клинических проявления между cиндромом Криглера Найяра I типа и синдромом Жильбера. Заболевание также обусловлено мутациями в кодирующей последовательности гена UGT1A1. Больные с СК-Н II часто являются компаундгетерозиготами, имеющими в одной хромосоме инсерцию в промоторе, а в другой – миссенс-мутацию в экзоне. Кроме того, описаны пациенты, несущие инсерции в промоторной области гена UGT1A1 в гомозиготном состоянии в сочетании со структурной мутацией в экзоне. В основе патогенеза СК-Н II – функциональная недостаточность фермента УДФ-ГТ1.
Манифестация заболевания наступает несколько позже, чем при I типе – от первых месяцев до первых лет. Как правило, больные доживают до юношества без каких-либо неврологических дефектов, хотя билирубиновая энцефалопатия может развиваться в более взрослом возрасте. Клинические проявления заболевания сходны с I типом, но менее тяжелые. Биохимические показатели: уровень билирубина крови менее 200 мкмоль/л. Гистологических изменений в печени не обнаруживают. Желчь пигментирована и содержит билирубин-глюкуронид. Проба с фенобарбиталом положительная
В Центре Молекулярной Генетики проводится прямая ДНК-диагностика данных синдромов, основанная на поиске мутаций во всех экзонах гена UGT1A1, анализе промоторной области.
Cиндром Криглера-Найяра
Синдром Криглера-Найяра относится к заболеваниям наследственного характера и выражается повышением содержания в крови непрямого (токсичного) билирубина. Синдром Криглера-Найяра может проявляться в двух вариантах.
Синдром I типа имеет злокачественное течение. У новорожденного появляются выраженные признаки желтухи, содержание билирубина в крови увеличивается, в результате чего наступает т.н. билирубиновая энцефалопатия. В первые два года жизни такие дети умирают. В редких случаях, пациент может дожить до подросткового возраста.
При синдроме II типа течение заболевания доброкачественное. Появление признаков желтухи может произойти только в подростковом возрасте или не появиться вообще.
Симптомы синдрома Криглера-Найяра
II тип синдрома может проявляться в виде желтухи в первые месяцы жизни или совсем не проявляться до достижения ребенком подросткового возраста. Течение заболевания доброкачественное и диагностировать его можно после специального обследования.
Лечение синдрома Криглера-Найяра
Лечение синдрома Криглера-Найяра направлено, прежде всего, на уменьшение содержания токсичного билирубина в крови и предупреждение развития повреждения головного мозга.
Методы лечения синдрома Криглера-Найяра
Интенсивность и методы лечения зависят от типа заболевания и тяжести его протекания. Предварительное тщательное обследование позволяет получить полную картину развития заболевания. Соответственно полученным данным разрабатывается стратегия лечения, включающая медикаментозное лечение, фототерапию, переливание крови и введение плазмы крови – все современные медицинские мероприятия, направленные на купирование патологического процесса и сохранение жизни и здоровья пациента.
Cемейные формы функциональных гипербилирубинемий в работе практического врача
Ребенок с синдромом Криглера — Найяра I типа Пациенты с функциональными гипербилирубинемическими состояниями длительное время (по нашим данным, от 6 месяцев до 3 лет и более) наблюдаются с первоначально ошибочными диагнозами. Таблица
 |
| Ребенок с синдромом Криглера — Найяра I типа |
Пациенты с функциональными гипербилирубинемическими состояниями длительное время (по нашим данным, от 6 месяцев до 3 лет и более) наблюдаются с первоначально ошибочными диагнозами.
Известно, что синдром желтух у детей представлен массой состояний. И если гемолитические, гепатитные и, реже встречающие в детском возрасте, механические (хирургические = подпеченочные) желтухи хорошо известны педиатрам и учитываются в дифференциальной диагностике, то так называемые семейные формы (функциональные гипербилирубинемические синдромы) чаще описываются в разделе казуистики [3, 4]. Но по мере изменения структуры заболеваемости на первый план выдвигаются наследственные синдромы. Пациенты с функциональными гипербилирубинемическими состояниями, прежде всего дети, длительное время (по нашим данным, от 6 мес. до 3 лет и более) наблюдаются с первоначально ошибочными диагнозами (гепатиты и т. п.). Между тем вспомнить о заболевании — это значит на 50% его диагностировать.
В связи с этим мы решили обобщить данные литературы и результаты собственных наблюдений.
По времени манифестации и тяжести проявлений одно из первых мест занимает синдром Криглера — Найяра [1].
Синдром Криглера — Найяра тип I — врожденная семейная негемолитическая желтуха с ядерной желтухой в результате полного отсутствия уридин-дифосфат-глюкоронилтрансферазы (УДФГТ) при нормальных функциях печени и отсутствии признаков гемолиза или резус-конфликта.
Патогенез заболевания заключается в отсутствии или резком снижении активности УДФГТ [6], в результате чего значительно повышается сывороточный уровень неконъюгированного жирорастворимого билирубина. Он связывается с альбумином, диффундирует через плацентарный и гематоэнцефалический барьеры (последнее — только у новорожденных). Связывающая (своеобразная буферная, компенсаторная емкость) альбумина уменьшается и соответственно клиническое течение синдрома ухудшается при гипоальбуминемии, ацидозе, увеличении концентрации органических ионов, назначении гепарина, салицилатов, сульфаниламидов, употреблении свободных жирных кислот. Проникновение несвязанного, неконъюгированного билирубина в клетки и митохондрии ведет к блокаде окислительно-фосфорилирующих реакций, особенно в гипоталамусе, хвостатом ядре, подкорковых ядрах, мозжечке.
| По частоте среди врожденных функциональных билирубинемий на первом месте стоит синдром Жильбера. Наш опыт работы в многопрофильных педиатрической и терапевтической клиниках показал, что в каждой из них за год выявляется порядка 10—20 случаев синдрома. |
Дифференциальная диагностика (ДД) проводится с синдромами Жильбера — Мойленграхта, Дабина — Джонса, Ротора, Люси — Дрисколла, ядерной желтухой новорожденных любой другой этиологии, врожденными циррозами печени и гепатитами, атрезией желчных ходов или тонкого кишечника (см. табл.).
Значительно благоприятней протекает синдром Криглера — Найяра типа II. Определение его [7] почти в точности повторяет синдром Криглера — Найяра типа I (врожденная семейная негемолитическая желтуха с ядерной желтухой при нормальных функциях печени), с той лишь разницей, что УДФГТ присутствует, хотя активность фермента значительно снижена. Уровень неконъюгированного билирубина может колебаться в пределах 60— 250 мг/л, фекального уробилиногена — 200—800 мг/л. Эффект от применения фототерапии и индукторов микросомальных ферментов хороший. Желтуха редко достигает степени ядерной, больные доживают до 50 и более лет, но в отделенном периоде, особенно при запоздалом лечении, нередки случаи глухоты, хореоатетоза, нейромышечных и личностых отклонений, гипоплазии зубов.
По частоте среди врожденных семейных функциональных билирубинемий на 1-м месте стоит синдром Жильбера. Наш опыт работы в многопрофильных — педиатрической и терапевтической — клиниках показал, что в каждой из них за год выявляется порядка 10 — 20 случаев синдрома. Но истинная частота, видимо, выше, поскольку диагностика заболевания строится главным образом на данных клиники и функциональной пробы с голоданием, а «клиническая мягкость» не привлекает внимание врача. Так, С. Д. Подымова [5] считает, что синдром Жильбера имеют не менее 1— 5% населения. Наблюдая таких больных, мы пришли к заключению, что синдром ассоциируется с генерализованной дисплазией соединительной ткани (особенно часто по типу синдромов Марфана и Элерса — Данлоса).
Патогенез синдрома неоднозначный. Обнаруживается небольшое снижение активности УДФГТ (на 10 — 30% по сравнению с нормой), однако основное значение придается нарушению захвата билирубина гепатоцитами. Последнее связывают с аномалией проницаемости мембран и с дефектом белка внутриклеточного транспорта. Эта гетерогенность клиренса билирубина проявляется также вариабельностью таких тест-субстратов, как бромсульфталеин и индоцианин зеленый.
Клинически для синдрома характерна субиктеричность различной степени выраженности. Чаще это выражается в иктеричности склер, матовой желтушности кожи, особенно лица, иногда частичном пожелтении стоп и ладоней, подмышечных впадин и носогубного треугольника. Нередко больные жалуются на общую слабость, подавленность, плохой сон, трудность концентрации внимания. Влияние голода на повышение содержания билирубина в сыворотке известно и для здоровых, но значительно выражено при синдроме Жильбера. С целью его выявления проводят пробу с голоданием. В течении 48 часов больной получает питание энергетической ценностью 400 ккал/сутки. В день начала пробы утром натощак и спустя 2 суток определяют билирубин сыворотки крови. При подъеме его на 50 — 100% проба считается положительной.
В последнее время многие специалисты полностью идентифицируют его с синдромом Мойленграхта, рассматривая их как одно состояние — синдром Жильбера — Мойленграхта — и определяют его как доброкачественную семейную конституциональную гибербилирубинемию в результате аномалии ферментов печени [7]. Большое значение для диагноза имеет семейный анамнез (легкая желтушность у членов семьи больного). Клиническая картина проявляется интермиттирующей мало выраженной желтухой, никогда не сопровождающейся ядерной. Желтуха усиливается при голодании, о чем говорилось выше, физических перегрузках, оперативных вмешательствах, употреблении алкоголя, инфекционных заболеваниях. Больных беспокоят тошнота, чувство переполнения желудка, тяжесть в эпигастрии, боли в правом подреберье, запоры или диспепсии. Почти у половины больных обнаруживается скрытый гемолиз (группа риска по холелитиазу!). Наряду с уже упомянутыми неврологическими расстройствами в рамках общей дисплазии соединительной ткани, свойственной этим больным, описывают и такие проявления, как пылающие и пигментные невусы, пигментация век, брадикардия и артериальная гипотония. Применение индукторов микросомальных ферментов очень эффективно. Прогноз хороший. Диагноз строится на клиническом исследовании и лабораторно-функциональных пробах. Гистологические находки (см. табл.) неспецифичны и обнаруживаются далеко не всегда. Поэтому ввиду общего нетяжелого состояния пациентов пункционная биопсия печени вряд ли оправданна (напомним, что риск обследования не должен превышать риска неустановленного диагноза и что прежде больные погибали от болезни, позже от лечения, а сейчас — от обследования).
ДД проводится со всеми типами неконъюгированной билирубинемии, синдромом Криглера — Найяра типа II, гемолитическими анемиями, шунтовыми гипербилирубинемиями (неэффективный гемопоэз с интрамедуллярным образованием значительного количества билирубина = талассемия, пернициозная анемия), постгепатической персистирующей гипербилирубинемией.
Синдром Дабина — Джонса — семейное нарушение выведения конъюгированного билирубина в желчные ходы, сочетающееся с отложением пигмента в печеночных клетках и умеренным увеличением печени («шоколадная печень»). При нем отсутствует нарушение глюкоронирования. Патогенез заключается в недостаточности транспорта конъюгированного билирубина внутрь и из гепатоцита. Билирубин попадает в кровь, его уровень в крови повышается, затем он усиленно выводится через почки. Заболевание может дебютировать в любом возрасте (разная пенетрантность гена?), но нередко проявляется после приема гормональных противозачаточных препаратов или при беременности.
| Влияние голода на повышение содержания билирубина в сыворотке известно и для здоровых людей, но значительно выражено при синдроме Жильбера. С целью его выявления проводят пробу с голоданием. В течение 48 часов больной получает питание энергетической ценностью 400 ккал/сутки. В первый день пробы натощак и спустя двое суток определяют билирубин сыворотки крови. При подъеме его на 50 — 100% проба считается положительной. |
Клинически ВВ проявляется в рецидивирующих периодах желтухи разной степени выраженности, но, как правило, не сопровождается зудом; увеличение размеров печени, а затем и селезенки умеренное. Функциональные печеночные пробы не изменены или изменены незначительно, однако наблюдается замедленное выведение контраста в желчный пузырь, повышение уровня копропорфирина I и снижение уровня копропорфирина III в моче. Гистологически в рибосомах печени откладывается меланиноподобный пигмент от желто-коричневого до черного цвета. Купферовские клетки остаются свободными, а соединительная ткань не разрастается.
ДД проводится со всеми формами конъюгированной и неконъюгированной гипербилирубинемии с желтухой: синдромы Жильбера — Мойленграхта, Ротора, Кароли (кистозное расширение внутрипеченочных желчных ходов), гепатиты, первичный билиарный цирроз, холестаз, синдром (симптомокомплекс) Циве. Синдром Циве — симптомокомплекс с гемолитической анемией, гиперлипидемией и желтухой, развивающийся у пациентов с неумеренным потреблением алкоголя. Он обусловлен увеличенным содержанием С16- и С18-жирных кислот в оболочке эритроцитов при уменьшении содержания длинноцепочечных ненасыщенных кислот. Провоцирующим фактором разрушения эритроцитов чаще является употребление алкоголя, после чего развиваются гемолиз и гиперлипидемия. Клинически синдром проявляется в основном после алкогольных эксцессов и сопровождается усиливающимися болями в правом подреберье и/или эпигастрии, лихорадкой, тошнотой, рвотой, потерей аппетита, увеличением размеров печени, а нередко и селезенки. При лабораторных исследованиях — повышение активности трансаминаз, щелочной фосфатазы, гамма-глютамилтрансферазы, анемия с манифестным или скрытым гемолизом, гиперхолестеринемия и/или гипертриглицеридемия. Гистологически — алкогольная жировая дистрофия печени с циррозом или без.
Формой синдрома Дабина — Джонса (или самостоятельной нозологической единицей?) является синдром Бюрка. При этом также обнаруживается липохромный гепатоз, но БЕЗ желтухи, хотя и СО ЗНАЧИТЕЛЬНОЙ гепатоспленомегалией.
Синдром Ротора — идиопатическая семейная доброкачественная гипербилирубинемия с адекватным повышением конъюгированного и неконъюгированного билирубина.
Патогенез заключается в нарушенном захвате неконъюгированного билирубина гепатоцитами, изменении его глюкоронирования и выведении с последующим рефлюксом билирубина в кровь.
Клинически синдром проявляется хронической желтухой (или субиктеричностью) кожи и слизистых. При этом ее интенсивность флюктуирует, увеличения размеров печени и селезенки не наблюдается. Гистологическая картина печени при световой микроскопии не изменена, при электронной микроскопии — митохондрии различных размеров, в фаголизосомах — пигментные тельца в виде решетчатообразных включений [8].
Диагнозы постгепатической доброкачественной гипербилирубинемии и физиологической желтухи новорожденных, постпрандиальных желтух (длительное употребление большого количества тугоплавких жиров), как прогностически благоприятные, строятся на принципе исключения более тяжелых вариантов гипербилирубинемий. Можно предположить, что подобные состояния связаны с маломанифестными ферментными аномалиями или аномалиями развития желчного пузыря (перетяжки, перегибы, особенно — в области сифона, сужения холедоха, приводящие к повышению давления в желчных ходах и избыточной нагрузке на гепатоцит).
| Синдром Дабина — Джонса может дебютировать в любом возрасте (разная пенетрантность гена?), но нередко проявляется после приема гормональных противозачаточных препаратов и при беременности. |
Семейный доброкачественный возвратный холестаз = синдром Аагенеса = Саммерскилла-Тигструппа = норвежский холестаз = холестаз с лимфедемами. Само обилие синонимов говорит о редкости синдрома: каждый вновь описываемый случай представляется авторам как открытие. Поскольку он редко встречается в литературе, определить истинную его распространенность весьма сложно. В основном он известен из описаний скандинавских авторов. Вероятно, патогенез заключается в гипоплазии лимфатических сосудов печени и других органов.
Клинически синдром проявляется как холестаз (наиболее чувствительный и специфический фермент холестаза — лейцин-аминопептидаза) уже в неонатальном периоде. Затем холестаз самостоятельно уменьшается, рецидивируя у взрослых. Медленно развиваются лимфатические отеки. Типичны гепатомегалия и нарушение функций печени. Гистологически он выражен внутрипеченочным холестазом с гигантоклеточной трансформацией.
Хронический холестаз сопровождается дефицитом жирорастворимых витаминов. Дефицит витамина Е приводит к прогрессирующей мозжечково-спинальной дегенерации (синдром Пермуттера).
ДД синдрома Аагенеса проводится со всеми синдромами холестаза и врожденных лимфатических отеков (синдромы Милроя, Вевера — Смита).
Злокачественный семейный холестаз (болезнь Байлера = синдром Клейтон — Юберга). В подавляющем большинстве случаев проявляется на 1-м году жизни [1]. Вначале холестаз (желтуха) самостоятельно разрешается через несколько недель или месяцев. Затем интенсивность желтухи нарастает, присоединяется мучительный зуд. Печень и селезенка существенно увеличиваются. В результате поражения паренхимы печени возникает геморрагический синдром. Эхографически печень выглядит плотной, напоминает цирротическую, в просвете желчного пузыря наблюдается густой застой. Прогноз плохой.
ДД проводят со всеми синдромами холестаза и синдромом Алажиль.
Синдром Люси — Дрисколл — аутосомно-рециссивно наследуемое редкое состояние, проявляющееся массивной гипербилирубинемией, развивающейся у всех детей, рожденных от одной матери страдающей этим заболеванием, в первые четыре дня их жизни.
Патогенез заключается в наличии фактора — ингибитора конъюгации билирубина в крови и моче матери и постнатально в крови и моче детей. Сыворотка таких матерей ингибирует in vitro конъюгацию билирубина в 5 — 7 раз сильнее, чем сыворотка женщин, не являющихся кондукторами данного синдрома. Вероятно, подобный фактор представлен стероидом, но он до сих пор не идентифицирован. Клиническая картина соответствует тяжелой гипербилирубинемии вплоть до ядерной желтухи.
ДД проводится с синдромами Криглера — Найяра типов I и II, новобиоциновой желтухой, эстрогеновой (транзиторная желтуха детей, вскармливаемых грудным молоком) и окситоциновой желтухой.
Лечение семейных функциональных гипербилирубинемий проводится по следующим принципам:
Литература
1. Алажиль Д., Одъевр М. Заболевания печени и желчных путей у детей. М.: Медицина, 1982. — 486 с.
2. Асоян А. В. Применение гипербарической оксигенации в комплексном лечении гипербилирубинемии у новорожденных с гемолитической болезнью. Автореф. дис. к. м. н. М., 1985. — 24 с.
3. Гасан Абу-Джабаль. Хронический гастродуоденит у детей на фоне дисплазии соединительной ткани. Автореф. дис. к. м. н. М., 1997. — 24 с.
4. Имамбаев С.Е. Эхография в диагностике заболеваний желчевыводящей системы у детей. Автореф. дис. к. м. н. М., 1986. — 24 с.
5. Подымова С. Д. Болезни печени. М.: Медицина, 1993. — 544 с.
6. Emery A., Rimoin D. (Ed.) Principles and practice of medical Genetics. London, Vv. 1, 2. — 1983.
7. Leiber B. Die klinischen Syndrome. Muenchen. — 1990.
8. Moelbert E., Marx R. Elektronenmikroskopiesche Untersuchungen am Lebergewebe beim Rotor-Syndrom. Acta hepato-splenoloy, 1966. — 13. — ss. 160 — 175.