Синдром корнелии де ланге что это
Синдром Корнелии де Ланге (синдром Брахмана де Ланге, синдром дегенеративного нанизма амстердамского типа) представляет собой редкое заболевание, впервые описанное Брахманом в 1916 г., но носящее имя датского педиатра де Ланге. Синдром считается очень редким, частота его составляет всего один случай на 100000 новорожденных (Beck, 1976).
а) Патогенез. Синдром Корнелии де Ланге сопровождается мутациями гена NIBPL короткого плеча пятой хромосомы (Gillis et al., 2004; Krantz et al., 2004; Tonkin et al., 2004). Недавно было выявлено, что описанные мутации является основным этиологическим фактором данного синдрома и выявляются у 27-56% пациентов (Yan et al., 2006).
Тем не менее, среди пациентов с синдромом Корнелии де Ланге выявляется ряд других хромосомных аномалий (Jackson et al., 1993). Проявления, позволяющие выявить наличие данного синдрома, отмечаются при частичной трисомии дистальной части 3-й хромосомы (3q21-3ter) и других перестройках 3-й хромосомы (DeScipio et al., 2005). Синдром дупликации 3q хромосомы (dup (3q)-синдром) только на первый взгляд имитирует синдром де Ланге (Holder et al., 1994).
Данные изменения и изменения, связывающие ген МЕСР2 с синдромом Ретта, доказывают, что следует соблюдать осторожность, рассматривая ген NIBPL как единственную причину развития синдрома де Ланге.
Синдром де Ланге может наследоваться доминантным путем; в действительности все случаи синдрома представляют собой вновь возникшие мутации. Риск повторного рождения ребенка с данной патологией составляет 2-5%.
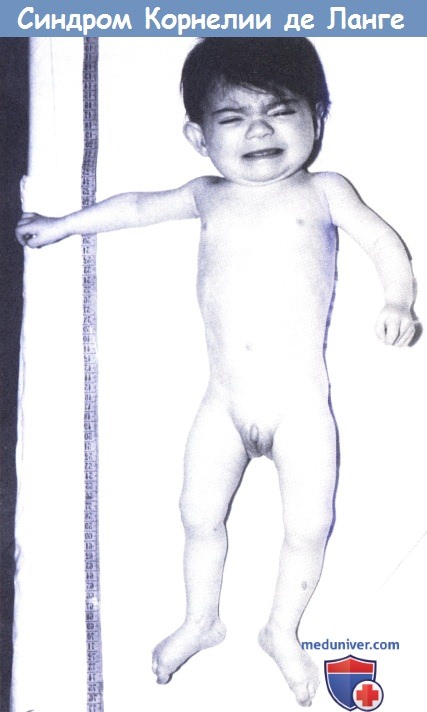 Синдром Корнелии де Ланге:
Синдром Корнелии де Ланге:
вдавленная переносица, низкая линия роста волос, сросшиеся брови,
короткие руки и согнутые пальцы рук и ног.
б) Клинические проявления. Синдром де Ланге является мультисистемным заболеванием, проявляющимся пре- и постнатальной задержкой роста, замедленным развитием, характерным дисморфизмом лица, мальформациями конечностей и множественными поражениями органов.
Фенотип характеризуется низкой массой при рождении, низким ростом, микроцефалией, генерализованным гирсутизмом и специфическим («любопытным») выражением лица со сросшимися бровями, низкой линией роста волос на лбу и шее, длинными ресницами, вдавленной спинкой носа, длинным подносовым желобком и вывернутыми ноздрями. Характерны плоские лопатообразные кисти и короткие конусовидные пальцы рук. Отмечается клинодактилия пятого пальца кисти.
Пальцы стоп располагаются проксимально, а возвышение большого пальца выражено незначительно. Могут встречаться более значимые аномалии конечностей, включая гипоплазию лучевой кости или уменьшение количества пальцев, часто одностороннее. Обычно отмечается микрогнатия (Hawley et al., 1985; Opitz, 1985). Зарегистрированы случаи атрофии зрительного нерва, колобомы зрительного нерва, проптоза и атрезии хоан. Часто встречается желудочно-кишечный рефлюкс, затруднения питания, пониженное слезоотделение и другие поражения глаз.
Кроме того, данное заболевание проявляется достаточно характерным поведенческим фенотипом, и в большинстве случаев серьезной или полной необучаемостью (Horsier и Oliver, 2006), часто сочетающейся с самодеструктивным поведением и избеганием социальных контактов, нередко достигая степени синдрома аутизма (Gillberg и Coleman, 2000; Arron et al., 2006; Bhuiyan et al., 2006).
Тем не менее, зарегистрированы редкие случаи пограничного или минимально нормального уровня интеллекта. По результатам работы двух исследовательских групп были выявлены значимые различия выраженности или пенетрантности некоторых фенотипов при наличии и отсутствии мутаций (Yan et al., 2006; Selicorni et al., 2007). Различные клинические проявления отмечаются и при разном характере мутаций (миссенс-мутациях и «усеченных мутациях»).
Среди пациентов с мутациями отмечается тенденция к более выраженным изменениям массы тела, роста и средней окружности головы при рождении, дисморфизму лица и нарушению речи, чем среди пациентов, у которых мутации отсутствуют.
в) Исход. В большинстве случаев тяжелые когнитивные и поведенческие нарушения сохраняются в течение всей жизни. Смертность повышена, частично вследствие нарушения функции желудочно-кишечного тракта в виде регургитации-рвоты-аспирации и развивающейся в тяжелых случаях смертельной пневмонии. Зарегистрированы отдельные случаи, когда пациенты доживали до 40 лет.
Редактор: Искандер Милевски. Дата публикации: 4.12.2018
Синдром Корнелии де Ланге – это генетически опосредованное мультисистемное заболевание, включающее множественные аномалии развития и олигофрению. Фенотипические признаки синдрома представлены микробрахицефалией, заниженной линией роста волос, тонкими сросшимися бровями, широкой запавшей переносицей, микрогенией и др. Характерна низкорослость, возможны врожденные пороки (ВПС, мочеполовые аномалии, пилоростеноз, диафрагмальная грыжа). При диагностике учитываются клинические критерии и результаты генетического тестирования. Дети с данной патологией нуждаются в симптоматической терапии, дефектологической помощи.
МКБ-10
Общие сведения
Синдром, характеризующийся комплексом стигм дизэмбриогенеза, пороками развития и умственной неполноценностью, был описан в 1916 г. доктором медицины В. Брахманом. Однако свое официальное название заболевание получило в честь педиатра из Амстердама Корнелии де Ланге, которая подробно описала сразу два клинических наблюдения в 1933 г. В литературе наряду с общепринятым – синдром Корнелии де Ланге – встречаются названия «синдром Брахмана де Ланге», «амстердамская карликовость/нанизм». Патология регистрируется с частотой 1:10000–1:30000, распространенность не имеет географических, расовых и гендерных различий.
Причины
Синдрому свойственна генетическая неоднородность, на данный момент известно три гена, ответственных за данную патологию. Около 50% описанных случаев связано с дефектами в гене NIPBL, кодирующем белок делангин (5p13.2), около 5% – с мутациями гена когезинового комплекса SMC1A (Xp11.22). Один известный случай ассоциирован с нарушением другой субъединицы когезина, кодируемой SMC3 (10q25). Примерно в 40% наблюдений синдром Корнелии де Ланге вызывается неизвестными мутациями, которые еще только предстоит вычислить.
Большая часть мутаций NIPBL являются вновь возникшими, поэтому чаще больные дети рождаются от генетически здоровых родителей. Однако известны наблюдения семейных случаев с аутосомно-доминантным наследованием. Ген SMC1A расположен на половой Х-хромосоме – при его дефектах наследование сцеплено с полом (болеют мужчины, женщины выступают гетерозиготными носительницами).
Факторы риска
Спонтанные патологические изменения генетического материала могут быть обусловлены следующими факторами:
При отсутствии генных мутаций у родителей риск повторного рождения ребенка с синдромом Брахмана де Ланге в одной семье равен 2-5%.
Патогенез
Молекулярную основу патологии составляет нарушение функционирования белков когезинового комплекса. Когезины играют важнейшую роль в процессе клеточного деления. Они располагаются внутри хромосомы, сцепляя и удерживая между собой две сестринские хроматиды. Другие, не менее важные функции когезина – это репарация ДНК и регуляция экспрессии генов.
По всей видимости, воздействие различных мутагенных факторов в критические периоды приводит к неправильному делению клеток и закладке органов. Патологоанатомическое исследование выявляет значительные изменения головного мозга: аплазию оперкулярной коры, недоразвитие роландовой борозды, запаздывающую миелинизацию и и миелодегенерацию. Также обнаруживаются пороки развития почек, надпочечников, гонад, сердца, гипоплазия тимуса.
Классификация
По степени выраженности фенотипических проявлений выделяют 2 варианта синдрома Корнелии де Ланге. Они различаются как проявленностью внешней симптоматики, так и витальным прогнозом:
Симптомы
Физическое развитие
Младенцы с синдромом Корнелии де Ланге рождаются с низкими росто-весовыми показателями: средняя масса составляет 2100-2300 г. Они малоактвны, редко выражают свои потребности криком. С первых дней отмечается вялое сосание груди, частые срыгивания, диспепсические расстройства. Такие проблемы нередко вынуждают кормить ребенка через назогастральный зонд или гастростому. Дети часто страдают рецидивирующими респираторными инфекциями. Физическое и психомоторное развитие отстает от возрастной нормы.
Фенотипические признаки
Внешний вид ребенка очень своеобразный. Перечисленные черепно-лицевые дисморфии входят в число основных критериев при диагностике патологии:
Скелетные аномалии
Пациенты имеют воронкообразную грудную клетку, короткую шею. В области позвоночного столба могут обнаруживаться такие дефекты, как Spina bifida, люмбализация или сакрализация позвонков. Присутствует гипоплазия кистей и стоп, дефекты развития пальцев (клинодактилия, синдактилия, олигодактилия), суставные контрактуры, дисплазия и врожденный вывих бедра. Отставание в росте сохраняется и во взрослом возрасте: мужчины с амстердамским нанизмом имеют рост около 156 см, женщины ‒ 131 см.
Неврологические и психопатологические проблемы
Больные с классической формой синдрома Корнелии де Ланге имеют интеллектуальные нарушения в степени имбецильности или глубокой дебильности. При мягкой форме интеллектуальный дефект выражен нерезко – больные имеют диагноз ЗПР. У четверти пациентов эпизодически отмечается судорожный синдром.
Поведенческие особенности представлены синдромом гиперактивности, тревожностью, признаками ОКР. Отмечается агрессивность по отношению к окружающим, аутоагрессия и самоповреждающее поведение, стереотипные действия. Речевые расстройства носят системный характер, страдает вербальная коммуникация, при небных расщелинах возникает ринолалия.
Зрительные нарушения
Распространенными проблемами со стороны зрительной системы выступают миопия, птоз века, блефарит. Реже диагностируется косоглазие, микрокорнеа, в единичных наблюдениях выявлены катаракта и глаукома. При тяжелом варианте синдрома Корнелии де Ланге высок риск отслойки сетчатки и атрофии зрительного нерва.
Пороки внутренних органов
Почти в половине случаев синдрому сопутствуют врожденные кардиальные пороки: дефекты перегородок сердца (аортолегочной, межжелудочковой, межпредсердной), коарктация аорты, тетрада Фалло. Аномалии развития ЖКТ чаще всего представлены гастроэзофагеальным рефлюксом (90%), может выявляться диафрагмальная грыжа (2%), пилоростеноз (4%), незавершенный поворот кишечника (10%). Частыми патологиями мочеполовой системы выступают поликистозная или подковообразная почка, гидронефроз, двурогая матка, неопущение яичек, гипоспадия. Характерна задержка полового развития.
Осложнения
Тяжелые варианты синдрома ассоциированы с пороками развития, несовместимыми с жизнью, и гибелью новорожденных вскоре после рождения. При более мягком течении тормозить развитие ребенка могут рецидивирующие синуситы, отиты с последующим развитием кондуктивной тугоухости. На фоне гастроэзофагеального рефлюкса нередко возникает анемия, аспирационная пневмония, стеноз пищевода, иногда − пищевод Барретта, аденокарцинома. В бытовом плане пациенты с синдромом Корнелии де Ланге нуждаются в уходе, всесторонней помощи. Им требуется специальное обучение и дефектологическое сопровождение.
Диагностика
При рождении диагностическая гипотеза о наличии у ребенка синдрома Корнелии де Ланге может быть выдвинута на основании визуально определяемых дефектов. Новорожденный должен быть осмотрен неонатологом, детским кардиологом, хирургом, неврологом. В плановом порядке проводятся консультации челюстно-лицевого хирурга, эндокринолога, ортопеда, логопеда. Диагностическая тактика включает:
Синдром Корнелии де Ланге: клиника, диагностика, лечение (случай из практики)
Полный текст:
Аннотация
Статья посвящена редкому синдрому с неясным ходом наследования. Описываются этиология, фенотипические признаки, симптомы, на основании которых педиатр и генетик могут заподозрить амстердамскую карликовость. Кратко изложены общие принципы лечения. Приведено клиническое наблюдение у ребенка Р. в возрасте 16 лет с полиорганным поражением.
Ключевые слова
Для цитирования:
Бугаенко О.А., Сиротченко Т.А., Бондаренко Г.Г., Вельковская М.М. Синдром Корнелии де Ланге: клиника, диагностика, лечение (случай из практики). Медицинский вестник Юга России. 2018;9(2):110-115. https://doi.org/10.21886/2219-8075-2018-9-2-110-115
For citation:
Buhayenko O.A., Sirotchenko T.A., Bondarenko G.G., Velkovchenko M.M. Syndrome of Cornelia de Lange. Medical Herald of the South of Russia. 2018;9(2):110-115. (In Russ.) https://doi.org/10.21886/2219-8075-2018-9-2-110-115
Введение
Первым сделал описание синдрома как самостоятельного заболевания немецкий врач В. Брахман в начале ХХ в. Несколько позже педиатр из Нидерландов Корнелия де Ланге (де Ланж) вела двух маленьких пациенток, страдающих этим заболеванием, и на материалах наблюдений подробно описала его. Эта патология может еще называться синдромом Брахмана де Ланге или дегенеративным нанизмом типа «Амстердам», т.к. трое детей с этим диагнозом жили в столице Нидерландов.
Заболевание встречается у новорожденных с частотой от 1:30000 до 1:10000, соотношение мальчиков и девочек — 1:1. Тип наследования не уточнен, известны семейные случаи с аутосомно-рецессивным типом наследования. У большинства больных кариотип нормальный. Синдром является генетически гетерогенным, обусловленным мутациями в гене NIPBL, SVC3, микродупликацией локусов q25-q29 хромосомы 3. Всего на данный момент известно более 400 случаев этого заболевания в разных странах [3][4].
В последнее время предполагают влияние на развитие данной патологии ряда следующих факторов риска:
В клинической картине синдрома выделяют следующие основные признаки:
Первые признаки заболевания визуально заметны уже у новорожденных. Кроме внешних особенностей, обращает на себя внимание маленький вес ребенка при рождении, который составляет 2/3 веса здорового ребенка, родившегося на аналогичном сроке беременности. Для новорожденных характерно наличие респираторного дистрегс-синдрома из-за специфического строения носоглотки, поэтому в анамнезе у этих пациентов регистрируются частые инфекционно-воспалительные заболевания дыхательных путей [6][7][8].
При вскрытии умерших больных обнаруживаются разнообразные дефекты головного мозга (недоразвитие нижней лобной извилины, расширение желудочков, дисплазия и гипоплазия извилин), гистология нередко показывает выраженную поперечную исчерченность нейронов внешнего зернистого слоя коры больших полушарий и расстройство топографии нейронов мозжечка.
Более чем в половине всех случаев амстердамскому нанизму сопутствуют дефекты в структуре сердца (аортолегочное окно; незаращенная перегородка, разделяющая как предсердия, так и желудочки, часто в комбинации с сосудистыми нарушениями; тетрада Фалло), дефекты в структуре ЖКТ (в основном, нарушения поворота кишечника), мочеполовой системы (кистозные образования почек, одиночные и множественные, иногда подковообразная почка и гидронефротические ее изменения, крипторхизм, двурогая матка) [9][10].
Это заболевание, характеризующееся множеством дефектов развития, является по своей сути пока еще не раскрытой генетической аномалией, которая начинается в период формирования эмбриона. Процесс, запущенный патогенным фактором, продолжается и усугубляется в дальнейшем, после рождения ребенка.
Выделяют два варианта синдрома: классический, со значительной задержкой физического и интеллектуального развития, грубыми пороками развития, и стертый, с лицевыми и малыми скелетными аномалиями, но пограничной задержкой психомоторного развития и отсутствием грубых пороков. Диагноз устанавливают на основании фенотипа, исследования кариотипа и методов цитогенетического анализа [2][6].
Специфического лечения не существует. Применяют нейрометаболические, ноотропные препараты, витамины, симптоматическую терапию, коррекцию логопедическую, психологическую.
Профилактика
Профилактикой синдрома, факторы возникновения которого точно не установлены заниматься сложно. Однако с учетом известных источников генных мутаций можно рекомендовать в качестве профилактических мер:
Прогноз
Продолжительность жизни зависит от многих факторов, главными из них можно назвать степень тяжести пороков жизненно важных органов, их раннюю диагностику и качество хирургических вмешательств по их ликвидации.
При аномалиях развития, несовместимых с жизнью, ребенок умирает на первой неделе жизни. В случае их незначительности или своевременного устранения хирургическим путем, больной с синдромом Корнелии де Ланге может прожить достаточно долго. Прогнозирование осложняется отсутствием сопротивляемости организма больных с данным синдромом ординарным, неопасным для обычных людей инфекциям, например, вирусным, которые тоже становятся причиной ранней смерти таких больных.
Средняя продолжительность жизни — примерно 1213 лет, по некоторым источникам больные со стертой формой заболевания или удачно проведенными операциями по устранению дефектов развития иногда доживали до пятого-шестого десятка лет [1][2].
Клинический случай
Опекуны больного Р., 15.10.2000 г.р., впервые обратились за помощью в республиканскую детскую больницу г. Луганска с жалобами на задержку физического и умственного развития у ребенка, снижение аппетита, периодические судорожные припадки, вздутие и увеличение в объеме живота.
Из анамнеза жизни больного известно, что мальчик родился от третьей беременности, протекавшей на фоне злоупотребления алкоголем, III срочных родов, с массой тела 2800 г., длиной — 52 см, окружностью головы — 33 см, окружностью груди — 32 см, оценкой по шкале Апгар — 8. Период новорожденности протекал на фоне затянувшейся физиологической желтухи. Ребенок находился на грудном вскармливании до 2-х месяцев, затем — на искусственном (коровье молоко). На первом году жизни часто болел простудными заболеваниями (ОРВИ), вследствие чего ребенок получал вакцинацию с отставанием от календаря прививок.
Мальчик проживал в социально-неблагополучной семье, где мать и отец злоупотребляли алкоголем и бродяжничали.
Неоднократные попытки участкового педиатра направить ребенка на обследование в ЦРБ к врачу-психиатру и невропатологу категорически отклонялись. В возрасте 1 года после осмотра детским неврологом был выставлен диагноз: гипоксически-ишемическое поражение ЦНС, перинатально обусловленное, гидроцефальный синдром, задержка психомоторного развития.
В 4-летнем возрасте хирургами диагностирована декомпенсированная врожденная патология кишечника (мегадолихоколон) на фоне дистрофии III степени и гипертрихоза. Из-за частых ОРВИ только через 6 месяцев произведена левосторонняя гемиколэктомия по Ребейну.
Проведены консультации специалистов.
Окулист: ОИ — спокойны, глазное дно — диски зрительных нервов деколорированы, четкие, сосуды обычные. (Примечание: деколорирование дисков зрительных нервов встречается при различных наследственных заболеваниях и отражает начинающуюся атрофию ДЗН).
Генетик: кариотипирование — набор хромосом 46ХУ, половой хроматин — 0 %. Заключение: синдром Корнелии де Ланге, задержка психического развития.
Эндокринолог: задержка физического развития, нанизм, вторичный гипогонадизм.
Логопед: общее недоразвитие речи II-III уровня.
Сурдолог: двусторонняя нейросенсорная тугоухость II-III ст.
Нефролог: неполное удвоение левой почки.
Кардиолог: вторичная миокардиодистрофия, СН I.
Невролог: детский церебральный паралич резидуального генеза, стойкая ремиссия, стойкие выраженные двигательные расстройства, грубая задержка моторного и психоречевого развития. Врожденная гидроцефалия. Эпилептический синдром резидуального генеза.
Хирург: Врожденная аномалия толстого кишечника, гипокинетический тип, субкомпенсированная форма (СПО 2005 год). Вторичный хронический колит.
Обсуждение
По результатам обследования установлен клинический диагноз: Синдром Корнелии де Ланге классическая форма. Задержка физического развития, нанизм генетически обусловленный. Задержка полового развития, вторичный гипогонадизм. Врожденная аномалия толстого кишечника, гипокинетический тип, субкомпенсированная форма (СПО 2005 г.). Вторичный хронический колит. Детский церебральный паралич, резидуального генеза, стойкая ремиссия, стойкие выраженные двигательные расстройства, грубая задержка моторного и психоречевого развития. Врожденная гидроцефалия. Эпилептический синдром резидуального генеза. Вторичная миокардиодистрофия, СН I. Неполное удвоение левой почки.
Главная задача терапии — увеличение продолжительности и улучшение качества жизни, дееспособности пациента, снижение проявлений симптоматики. Пациенту проводилась коррекция противосудорожными препаратами (депакин, финлепсин) миорелаксантами (мидокалм), ноотропными препаратами (фенибут), метаболическими средствами (рибоксин, аспаркам), ферментными препаратами (панкреатин), пробиотиками, гепатопротекторами, поливитаминами, анаболическими стероидами. Также назначался комплекс лечебной физкультуры, массаж.
В катамнезе в течение 2 лет наблюдения: ремиссия фебрильных судорог — 30 месяцев, частые острые респираторные инфекции до 6 раз в год, снижение слуха. Прогрессирование когнитивной недостаточности не отмечается.
Заключение
Приведенный клинический случай подтверждает необходимость проведения комплексного обследования детей с задержкой физического, псхоречевого и полового развития. Орфанные заболевания являются мультидисциплинарной проблемой, так как в патологический процесс вовлечены несколько органов и систем. Своевременная диагностика, лечение и коррекционные мероприятия позволят уменьшить клинические проявления, улучшить качество жизни и социальную адаптацию пациентов.
Список литературы
1. Козлова С.И. Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, Авторская академия; 2007.
3. Гинтер Е.К. Медицинская генетика. – М.: Медицина; 2003.
4. Жимулев И.Ф. Общая и молекулярная генетика. – Новосибирск: Сиб. унив. изд-во; 2002.
5. Бородулин В.И., Тополянский А.В. Синдромы и симптомы в клинической практике: эпонимический словарьсправочник. – М.: Эксмо; 2009.
6. Berney T.P., Ireland M., Burn J. Behavioral phenotype of Cornelia de Lange syndrome. // Arch. Dis. Child. – 1999. – Vol. 81. – P. 333-336.
7. Deardorff M.A., Krantz I.D. Cornelia de Lange Syndrome // Encyclopedia of Neuroscience. – 2009. – P. 159-162. doi: org/10.1016/b978-008045046-9.01491-1.
8. Badoe E.V. Classical Cornelia de Lange syndrome. // Ghana Medical Journal. – 2010. – Vol. 40, issue 3. – P. 148-150
10. Белозеров Ю.М. Детская кардиология (наследственные синдромы). – Элиста: ЗАОр «НПП «Джангар»; 2008.
Об авторах
Бугаенко Оксана Александровна – к.м.н, ассистент, кафедра педиатрии факультета последипломного образования.
Сиротченко Тамара Анатольевна – д.м.н, профессор, кафедра педиатрии факультета последипломного образования.
Бондаренко Галина Григорьевна – к.м.н, доцент, кафедра педиатрии факультета последипломного образования.
Вельковская Мария Марковна – врач-инфекционист.
Для цитирования:
Бугаенко О.А., Сиротченко Т.А., Бондаренко Г.Г., Вельковская М.М. Синдром Корнелии де Ланге: клиника, диагностика, лечение (случай из практики). Медицинский вестник Юга России. 2018;9(2):110-115. https://doi.org/10.21886/2219-8075-2018-9-2-110-115
For citation:
Buhayenko O.A., Sirotchenko T.A., Bondarenko G.G., Velkovchenko M.M. Syndrome of Cornelia de Lange. Medical Herald of the South of Russia. 2018;9(2):110-115. (In Russ.) https://doi.org/10.21886/2219-8075-2018-9-2-110-115
Синдром корнелии де ланге что это
Эту группу заболеваний составляет ряд синдромов, хотя и относимых к генетически обусловленным, но встречающихся главным образом спорадически. Однако описания монозиготных близнецов с данной патологией всегда свидетельствуют об их конкордантности; кроме того, иногда встречаются семейные случаи этих заболеваний.
Вместе с тем, насколько позволяет судить анализ значительных выборок этих синдромов, наследование здесь не имеет характера простой менделевской передачи мутантного гена. Часть этих синдромов, возможно, окажется при более совершенном цитогенетическом исследовании хромосомной патологией. Так, уже в последние годы выявлены микроделеции при ряде заболеваний, ранее относимых к этой группе (синдром «лицо эльфа» и др.).
Приводим описание лишь наиболее распространенных из этих синдромов.
Синдром Корнелии де Ланге (амстердамская карликовость). Впервые синдром описан в Голландии педиатром К. de Lange в 1933 г. у 2 девочек из неродственных семей. К настоящему времени имеются описания более 400 больных в различных странах.
Данные о популяционной частоте синдрома значительно варьируют от 1:30 000 рождений до 1:10 000-12 000 [Leavitt A. et al., 1985; Opitz G. et al., 1985].
Клиническая картина заболевания в типичных случаях весьма характерна. Длина и масса тела больных значительно отстают от нормы. Отмечается выраженное своеобразие в строении лица: густые сросшиеся брови, длинные густые загнутые ресницы, короткий нос с развернутыми вперед ноздрями и вдавленной переносицей, увеличенное расстояние между основанием носа и верхней губой, тонкие губы с опущенными углами рта. Череп уменьшен, брахицефальной структуры. Характерны аномалии строения верхних конечностей: кисти небольших размеров, короткий II палец и проксимально расположенный I палец, искривленный V. Нередко отмечается синдактилия стоп (И— III, III— ГУ пальцы). Наблюдаются и другие аномалии конечностей и суставов: уменьшение числа пальцев, сгибательные контрактуры, а также деформация позвоночника и грудины. Очень часто бывают различные пороки внутренних органов, особенно аномалии строения почек [Лазюк Г. И. и др., 1983; Jackson L. et al., 1993].
На коже у больных, кроме гипертрихоза, резко выраженного в области спины и поясницы, нередко отмечается общая мраморность, характерны краснота кончика носа, цианоз носогубной области.
Среди неврологических нарушений, как правило, наблюдаются мышечная гипотония, оживление сухожильных рефлексов, но возможен и гипертонус с мышц конечностей.
Диагноз синдрома Корнелии де Ланге не всегда прост, так как встречаются умственно отсталые дети с небольшим числом аномалий, входящих в данный синдром. При отсутствии несомненного биологического метода диагностики трудно сказать, можно ли такие случаи относить к синдрому Корнелии де Ланге. Как известно, этот сложный вопрос касается и других заболеваний, диагностика которых осуществляется на основе только клинического своеобразия.
Умственная отсталость определяется практически у всех больных с данным синдромом: в 80 % случаев устанавливают имбецильность или глубокую дебильность. Однако описаны и больные с нерезко выраженным интеллектуальным дефектом (IQ = 73-75).
В литературе есть указания на то, что у больных с данным синдромом имеются стремление к аутоагрессии и склонность к стереотипным движениям — бегу по кругу, вращению, стереотипным движениям руками [Moore M, 1970]. Судорожный синдром отмечается у 25 % больных: судороги, как правило, только эпизодические, а не в виде частых и полиморфных приступов.
При рентгенографии черепа нередко обнаруживаются явления внутричерепной гипертензии. При электроэнцефалографии каких-либо специфических, характерных для синдрома изменений не выявляется.
Патологическая анатомия при синдроме Корнелии де Ланге изучена достаточно подробно [Лазюк Г. И. и др., 1974; Лурье И. В., Черствая Е. И., 1979]. Как наиболее характерное нарушение мозга описывается двусторонняя аплазия оперкулярных отделов лобных долей. Отмечают также отсутствие центральной (роландовой) борозды, гипоплазию пирамид обонятельного нерва, верхних височных извилин, задней спайки мозолистого тела, отставание миелинизации и распад миелина, глиоз и очаговую аплазию клеток наружного зернистого и пирамидного слоев во всех отделах мозга.
Этиология синдрома остается неясной. Несомненно, поражение возникает в ранние сроки эмбрионального развития. Описано значительное число случаев синдрома Корнелии де Ланге с различными хромосомными нарушениями, однако роль их неясна [Opitz J., 1985]. Наряду с этим наблюдались семьи с пораженными сибсами (без хромосомной патологии), что позволяет некоторым авторам высказывать мнение об аутосомно-рецессивном наследовании синдрома [Блюмина М. Г, 1971]. Известны также случаи, когда заболевание передается от родителей, имеющих неглубокий дефект и соматические черты синдрома [Blank С., 1985; Leavitt A. et al., 1985].
Лечение. Специфического лечения не существует. При необходимости проводят противосудорожную и седативную терапию. Применяют ноотропы, анаболические гормоны (неробол, ретаболил), назначают витаминотерапию.
Синдром Рубинштейна — Тейби впервые описан в 1963 г. у 7 умственно отсталых детей. Его второе название «синдром широкого I пальца кисти и стопы с лицевыми аномалиями». Точных данных о частоте синдрома нет. И. В. Лурье (1983) приводит ориентировочные сведения о частоте заболевания у новорожденных, равной приблизительно 1:25 000— 30 000.
Клиническая картина характеризуется умственной отсталостью, задержкой роста и специфическими особенностями строения лица и тела, основные из которых — короткий и широкий I палец на кисти и стопе, своеобразное лицо с длинным загнутым носом, антимонголоидным разрезом глаз, гипертелоризмом, недоразвитием верхней челюсти, брахицефальной структурой черепа. У больных низкий рост волос на лбу, иногда спускающихся в центре утлом. Кроме расширения концевой фаланги I пальца, отмечается ее отклонение с искривлением в межфаланговом суставе. Иногда бывают расширение концевых фаланг других пальцев, синдактилия и полидактилия стоп, косолапость, врожденный вывих бедра, повышенная разгибаемость суставов. Повышена склонность к катаральным состояниям дыхательных путей и пневмониям, а также к гнойничковым заболеваниям кожи.
Характерная для больных задержка роста происходит в основном постнатально. Масса тела при рождении почти не снижена. Часто встречаются различные врожденные пороки внутренних органов — сердца, мочеполовой системы, органов дыхания, диафрагмальная грыжа. Весьма типична для синдрома патология глаза: катаракты, колобомы, аномалии рефракции, глаукома, нистагм, атрофия зрительных нервов, косоглазие, заращение слезно-носового канала.
При рентгенографии выявляются микроцефалия, укорочение черепных ям, иногда длительное незарастание большого родничка, симптомы открытой гидроцефалии.
Дерматоглифика всегда изменена, но неспецифична.
Умственная отсталость носит характер интеллектуального недоразвития различной степени; как правило, она довольно глубокая, но описаны также случаи пограничного снижения интеллекта (IQ = 70—80) и даже синдрома с нормальным интеллектом [Pratesic et al., 1972]. Чаще всего интеллектуальное недоразвитие соответствует олигофрении в степени имбецильности. Судорожный синдром встречается у 20—25 % больных, что соответствует его частоте среди детей с выраженной умственной отсталостью [Пурас Д. К., 1988, и др.]. Иногда у больных наблюдаются склонность к агрессивным реакциям, аутоагрессивное поведение, частые аффективные вспышки.
Анатомически из пороков развития мозга наиболее часто встречается агенезия мозолистого тела. Обнаруживают также обеднение слоев коры пирамидальными клетками и задержку темпов миелинизации. У 25 % больных находят различные пороки внутренних органов.
Этиология синдрома остается неясной. Оба пола поражаются с одинаковой частотой. В последние годы появились работы, указывающие на роль микроделеции хромосомы 16, но не во всех случаях [Jones К., 1997].
Лечение. Специфического лечения не существует. Часто больные нуждаются в различных хирургических операциях. Проводят неспецифическую терапию, как и при других формах умственной отсталости.
Умственная отсталость с гипертрихозом. Данная форма интеллектуального недоразвития впервые была описана у 9 детей обоего пола Г. С. Марничевой и соавт. в 1976 г.
Среди контингента больных, наблюдавшихся медико-генетической консультацией, этот синдром встречается приблизительно с той же частотой, что и синдром Корнелии де Ланге. С равной частотой поражаются оба пола.
Клиническая картина. Одним из наиболее характерных соматических признаков является гипертрихоз, который отмечается уже при рождении ребенка. Гипертрихоз выражен главным образом на спине, на разгибательной поверхности конечностей, имеются также значительное пушковое оволосение на лбу, щеках, длинные густые ресницы. Лицо больного бледное, пастозное, с несколько отечными веками, широким носом, антимонголоидным разрезом глаз, толстой оттопыренной нижней губой. Верхняя губа особой формы, с опущенным в центре углом. Нижняя челюсть несколько недоразвита, подбородок уменьшен. Почти у всех детей радужная оболочка имеет своеобразный густой синий цвет. Зубы мелкие и редкие. У больных широкие, крупные конечности, короткая шея, фигура массивная. Кисти рук большие, с широкими концевыми фалангами
Признаки нарушенного внутриутробного развития: крипторхизм, деформация ушных раковин, искривление костей предплечья, врожденная косолапость, наличие сосудистых, депигментированных и пигментных пятен и др. Нередки аномалии глаз: частичная атрофия зрительных нервов, врожденный нистагм, глиоз дисков зрительных нервов, врожденные аномалии сосудов сетчатки (извитость), выраженная дальнозоркость или резкая миопия.
У больных повышена склонность к респираторным заболеваниям, хроническому риниту, гаймориту, аллергическому конъюнктивиту. Для них с рождения характерны общая вялость, мышечная слабость, плохой аппетит, бледность, анемичность и пастозность (на этом основании иногда больным ошибочно ставится диагноз гипотиреоза).
Неврологические симптомы проявляются понижением мышечного тонуса, у некоторых детей со снижением сухожильных рефлексов. Мышечная гипотония накладывает отпечаток на выражение лица: опущенные щеки, тяжелые веки, оттопыренная нижняя губа. Наблюдается также моторная неловкость, медлительность. У некоторых детей были нерезкие явления атаксии, очень часто слюнотечение. Судорожный синдром наблюдается редко, как правило, лишь на высоте лихорадки. Встречаются редкие приступообразные состояния без потери сознания с резкой бледностью, вялостью, обездвиженностью.
Различна глубина интеллектуального дефекта Встречаются случаи как очень тяжелой олигофрении, так и пограничной умственной отсталости.
Очень характерны для рассматриваемого синдрома расстройства речи. Поэтому он назван «умственная отсталость с гипертрихозом и речевым недоразвитием». Речевые нарушения выражены больше, чем обычно при олигофрении. Иногда речевой дефект является ведущим симптомом в психическом статусе (особенно при неглубокой интеллектуальной недостаточности). В связи с этим таким больным нередко ставится диагноз моторной алалии (настолько четко выражено расхождение между речевым недоразвитием и общим интеллектуальным уровнем). Однако в большинстве случаев речевые нарушения имеют характер дизартрии. Уже в раннем возрасте у некоторых детей отмечаются очень тихий крик, неумение зевать, смеяться, что свидетельствует о поражении периферического речевого аппарата. В других, более легких случаях на первый план выступают слабая речевая активность, недостаточная автоматизация речи при логопедических занятиях при отсутствии дизартрии, т. е. задержка речевого развития. Всем детям свойственны вялость, аспонтанность, медлительность, а также выраженные колебания психического тонуса, что в более старшем возрасте резко сказывается на работоспособности.
Трудностей поведения при этом синдроме обычно не наблюдается. Такие дети бывают чаще всего беззащитны и не могут за себя постоять в детском коллективе.
Заслуживает внимания тот факт, что детей с синдромом гипертрихоза объединяет не степень интеллектуальной недостаточности, а ее структура и дополнительные психопатологические особенности. Если по степени интеллектуального недоразвития больные распределялись от пограничной умственной отсталости до выраженной имбецильности, то речевые и личностные нарушения с неизменным постоянством были отмечены у всех больных.
Дифференциальная диагностика основана на отграничении поражений с наиболее типичными при данном синдроме соматическими изменениями. Учитывая то, что постоянным и самым ярким признаком синдрома считается первичный, отмечавшийся с рождения, так называемый ланугинозный гипертрихоз, дифференциальная диагностика проводится прежде всего с теми заболеваниями, которые сопровождаются первичным гипертрихозом. Все варианты гипертрихоза, сочетающегося с преждевременным появлением вторичных мужских половых признаков, исключаются, поскольку этих симптомов не отмечалось ни у одного из наблюдавшихся нами детей. Гипертрихоз имеется также при некоторых известных формах олигофрении: синдромах Корнелии де Ланге, Рубинштейна — Тейби, при трисомии-18, мукополисахаридозах. Дифференциальная диагностика с этими заболеваниями не вызывает затруднений, так как все они сопровождаются специфическими для каждой формы соматическими признаками, иными, чем при данном синдроме.
Основными, опорными для диагноза симптомами этого заболевания являются первичный общий гипертрихоз и своеобразное лицо. Вспомогательную роль в диагностике играют другие соматические изменения, а также однотипная структура умственной отсталости с выраженным речевым недоразвитием.
Этиология и патогенез. Этиология заболевания остается неясной. Все наблюдавшиеся случаи (более 80) были спорадическими. Как правило, психически здоровыми были родители и сибсы, а больные были интеллектуально полноценными. Проведенные цитологические исследования пока не дают основание полагать, что данный синдром может быть хромосомным заболеванием.
Не выявлено также каких-либо однотипных биохимических изменений, которые позволили бы говорить о наследственном дефекте обмена. В анамнезе не было никакого экзогенного фактора, способного оказать тератогенное воздействие.
Данное поражение, несомненно, развивается внутриутробно. Это подтверждается наличием таких признаков, как врожденный гипертрихоз и накопление у больных внутриутробно обусловленных аномалий развития.
Соматические изменения при данном синдроме могут свидетельствовать о нарушении регуляции многих функций гипоталамуса: водного обмена (отечность), устойчивости к инфекции, часто отмечаются аллергические состояния, постоянными симптомами являются вялость и аспонтанность. Наконец, гипертрихоз является следствием нарушения механизма эндокринной регуляции, хотя в данном случае все это остается неясным. Первичной дисфункции щитовидной железы не выявлено.
Лечение должно быть направлено на стимуляцию развития речи и моторики. Отмечен положительный эффект от применения препаратов стимулирующего действия (церебролизин, витамины группы В, аминалон, ноотропы, сиднокарб и др.).
Необходимы также массаж и лечебная физкультура. Больным с выраженной заторможенностью, вялостью могут быть рекомендованы небольшие дозы тиреоидина. Состояние детей с более легким нарушением существенно улучшалось при логопедических занятиях.