Синдром Клиппеля-Фейля
М. Klippel и A. Feil описали врожденный порок развития с характерной триадой симптомов: короткая шея, низкорасположенная граница волос на шее, ограничение подвижности головы.
Что провоцирует / Причины Синдрома Клиппеля-Фейля:
Аномалия развивается в результате нарушения васкуляризации, сегментации, аплазии, гипоплазии, задержки слияния парной закладки позвонков в эмбриональном и фетальном периодах. Сформировавшиеся синостозы шейных, а иногда и верхних грудных позвонков, уменьшение числа шейных позвонков до 4-5, несращение дуг и тел позвонков определяют клиническую картину и тяжесть деформации при данном синдроме.
Симптомы Синдрома Клиппеля-Фейля:
Классическая триада симптомов при синдроме Клип-пеля-Фейля включает короткую шею, низкорасположенную границу волос на шее и ограничение подвижности головы. Наиболее выраженным симптомом является укорочение шеи, но в плане диагностики наиболее информативным симптомом является ограничение движений головы. В большей степени страдают ротационные движения.
Скрытые аномалии развития внутренних органов у больных с этим заболеванием представляют большую угрозу для жизни больного, чем сам синдром. У 35% больных имеются тяжелые аномалии развития почек в виде аплазии, гипоплазии, гипоплазии лоханок, гидронефроза, эктопии мочеточников. Довольно часто встречаются аномалии развития сердечно-сосудистой системы [дефекты межжелудочковой перегородки, незаращение артериального (боталлова) протока, декстропозиция аорты, отсутствие легкого и др. ].
Рентгенологическая картина
На основании рентгенологического метода исследования по Фейлю различают два типа заболевания.
Второй тип заболевания по Фейлю характеризуется наличием синостоза атлантозатылочного сустава, костными сращениями нижележащих позвонков, шейных ребер. Аномалия шейного отдела позвоночника сочетается с неврологическими расстройствами.
Для выявления нестабильности позвонков, сужения позвоночного канала, гипоплазии и аплазии дисков, гипермобильности суставов рентгенограммы шейного отдела позвоночника в сагиттальной плоскости выполняют в положении максимального сгибания и разгибания головы.
Лечение Синдрома Клиппеля-Фейля:
Прогноз
Для функции неблагоприятный, сохраняется ограничение движений головы. Вторичные дегенеративные изменения позвоночника в редких случаях приводят к тяжелым неврологическим расстройствам.
К каким докторам следует обращаться если у Вас Синдром Клиппеля-Фейля:
Синдром клиппеля фейля что это такое
а) Терминология:
1. Синонимы:
• Синдром Клиппеля-Фейля
2. Определения:
• Врожденная мальформация позвоночника, характеризующаяся нарушением сегментации ≥ двух шейных позвонков ± аномалии сегментации грудного и поясничного отделов позвоночника
1. Общие характеристики аномалии Клиппеля-Фейля:
• Наиболее значимый диагностический признак:
о Одно- или многоуровневые врожденные аномалии сегментации и слияния шейных позвонков
• Локализация:
о С2-3 (50%) > С5—6 (33%)>КВС, верхнегрудной отдел
• Размеры:
о Тела позвонков уменьшены в размерах, на уровне межтелового пространства принимают форму усеченного конуса
о Рудиментарное межтеловое пространство отличается небольшими высотой и диаметром
• Морфология:
о Сужение тела позвонка («осиная талия») на месте рудиментарного межтелового пространства ± сращение задних элементов
3. КТ признаки аномалии Клиппеля-Фейля:
• Костная КТ:
о Типичные костные признаки ± дегенеративные изменения
о Сагиттальный и поперечный размеры спинномозгового канала обычно остаются нормальными
— Сужение спинномозгового канала свидетельствует о дегенеративных изменениях смежных с блокированными сегментов
— Расширение канала → скорее всего связано с сирингомиелией
4. МРТ признаки аномалии Клиппеля-Фейля:
• Т1-ВИ:
о Сращение шейных позвоночно-двигательных сегментов: тел позвонков ± дугоотростчатых суставов, задних элементов
о ± дегенеративные изменения: спондилез, грыжи межпозвонковых дисков наблюдаются достаточно часто (особенно на нижнешейном уровне)
о ± костные аномалии КВС, мальформация Киари 1
• Т2-ВИ:
о Костные изменения аналогичны таковым на Т1-ВИ, нормальный сигнал костного мозга
о ± компрессия спинного мозга или корешков, сирингомиелия, аномалии ствола мозга, миелошизис
5. Рекомендации по проведения исследования:
• Наиболее оптимальный метод диагностики:
о Рентгенография для оценки протяженности изменений, выявления нестабильности, дегенеративных изменений
о МРТ для исключения компрессии спинного мозга, диагностики дегенеративных изменений
• Протокол исследования:
о Рентгенограммы в нейтральном положении и в положении сгибания/разгибания в динамике для диагностики прогрессирующей нестабильности, дегенеративных изменений
о Многоплоскостная МРТ для оценки степени стеноза спинномозгового канала, компрессии спинного мозга, дегенеративных изменений мягких тканей
о УЗИ или КТ с КУ для диагностики и определения тяжести сочетанных аномалий внутренних органов
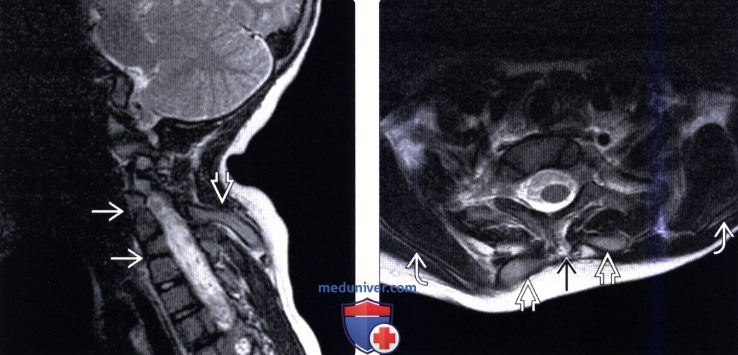 (Слева) Т2-ВИ (СКФ тип 1, лопаточно-позвоночные кости с обеих сторон): признаки аномального сглаживания лордоза шейного отдела позвоночника и множественные аномалии сегментации позвонков. Лопаточно-позвоночная кость располагается в толще мягких тканей кзади от позвоночника и сочленяется с задними элементами позвонков.
(Слева) Т2-ВИ (СКФ тип 1, лопаточно-позвоночные кости с обеих сторон): признаки аномального сглаживания лордоза шейного отдела позвоночника и множественные аномалии сегментации позвонков. Лопаточно-позвоночная кость располагается в толще мягких тканей кзади от позвоночника и сочленяется с задними элементами позвонков.
(Справа) На аксиальном Т2-ВИ (СКФ тип 1) с обеих сторон от позвоночника видны лопаточно-позвоночные кости, соединяющие задние элементы позвонков с лопатками. 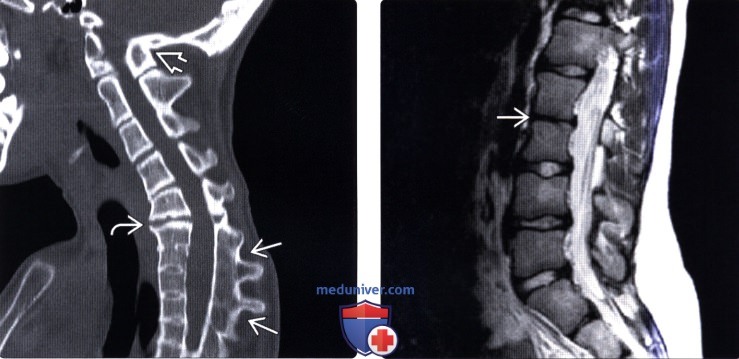 (Слева) Сагиттальный КТ-срез шей но- грудного отдела позвоночника: отсутствие сегментации шейных и грудных позвонков. Обратите внимание на остистый отросток С1, сросшийся с затылочной костью, и сращение тел и задних элементов С6-Т4. Обратите также внимание на быстро формирующиеся дегенеративные изменения межпозвонкового диска С5-С6 сегмента, который в данном случае блокирован не был.
(Слева) Сагиттальный КТ-срез шей но- грудного отдела позвоночника: отсутствие сегментации шейных и грудных позвонков. Обратите внимание на остистый отросток С1, сросшийся с затылочной костью, и сращение тел и задних элементов С6-Т4. Обратите также внимание на быстро формирующиеся дегенеративные изменения межпозвонкового диска С5-С6 сегмента, который в данном случае блокирован не был.
(Справа) Сагиттальное Т2-ВИ (СКФ тип 3) подтверждает наличие аномалии сегментации поясничного отдела позвоночника, аналогичные аномалии имеют место на уровне шейного и грудного отделов позвоночника (здесь не показаны). 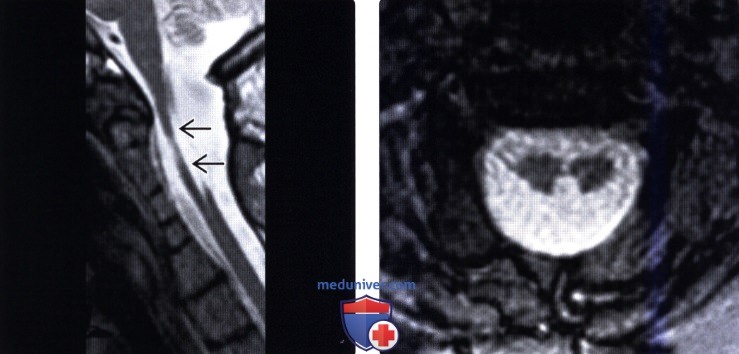 (Слева) На сагиттальном Т2-ВИ (СКФ тип 1) показан случай тяжелого многоуровневого нарушения сегментации шейного отдела позвоночника, тела позвонков и межпозвонковые диски с признаками гипоплазии, кроме того отмечается хорошо заметное истончение спинного мозга, на аксиальных томограммах в этом случае выявлена диастематомиелия с расщеплением спинного мозга на две примерно равные по размерами части.
(Слева) На сагиттальном Т2-ВИ (СКФ тип 1) показан случай тяжелого многоуровневого нарушения сегментации шейного отдела позвоночника, тела позвонков и межпозвонковые диски с признаками гипоплазии, кроме того отмечается хорошо заметное истончение спинного мозга, на аксиальных томограммах в этом случае выявлена диастематомиелия с расщеплением спинного мозга на две примерно равные по размерами части.
(Справа) Аксиальная T2*GRE MPT (СКФ тип 1) подтверждает наличие сочетанной аномалии — диастематомиелии шейного отдела спинного мозга (тип 2, отсутствие перегородки).
в) Дифференциальная диагностика аномалии Клиппеля-Фейля:
1. Ювенильный идиопатический артрит:
• Сложно дифференцировать с костным блоком на уровне шейных позвонков
• Обращайте внимание на поражения других суставов, особенности анамнеза заболевания
2. Послеоперационный костный блок:
• Отсутствие «талии» на уровне межтелового пространства, анкилоз дугоотростчатых суставов наблюдается нечасто
• Ключом к постановке диагноза является хирургическое вмешательство в анамнезе
3. Отдаленные последствия дисцита:
• Неровные края замыкательных пластинок, отсутствие «талии», ± кифотическая деформация
• Диагноз подтверждается наличием в анамнезе указаний на перенесенную инфекцию позвоночника
4. Анкилозирующий спондилит:
• Тонкие сливающиеся друг с другом (непрерывно идущие вдоль позвоночника) синдесмофиты («бамбуковая палка») + симметричное поражение крестцово-подвздошных суставов
• Положительный тест на HLA-B27 (95%)
1. Общие характеристики:
• Этиология:
о Общепринятого мнения об этиологии заболевания не существует; считается, что причиной может быть воздействие тех или иных факторов между четвертой и восьмой неделями эмбрионального развития:
— К таким причинным факторам относят воздействие тератогенов, алкоголизм матери
— Сочетание с другими синдромами (алкогольный синдром плода, синдром Голденгара, синдром Вильдерванка (шейно-окуло-акустический синдром))
• Генетика:
о Заболевание регистрируется спорадически; у многих пациентов отмечена семейная генетическая предрасположенность с различной экспрессивностью патологического признака
— Сращение С2/3 (тип 2) → аутосомно-доминантный тип наследования с вариабельной пенетрантностью
— Сращение С5/6 (тип 2) → аутосомно-рецессивный тип наследования
о Ген SGMI (8 хромосома) → первый идентифицированный ген, ответственный за развитие синдрома Клиппеля-Фейля; экспрессия этого гена наблюдается при всех трех типах СКФ
• Сочетанные аномалии:
о Полупозвонки, позвонки-»бабочки», spina bifida
о Сколиоз (обычно врожденный) ± кифоз (60%)
о Дисплазия зубовидного отростка, базилярная импрессия, ассимиляция С1, шейно-затылочная нестабильность
о Сирингомиелия, диастематомиелия (20%), мальформация Киари 1 (8%), нейроэнтеральная киста или дермоид (редко)
о Нейрошизис шейного отдела позвоночника ± синкинезия (20%)
о Деформация Шпренгеля±лопаточно-позвоночная кость (1 5-30%), одно- или двусторонняя
о Нейросенсорная тугоухость (30%), аномалии наружного уха, аномалии мочеполовой системы (35%), врожденные пороки сердца (14%), деформации верхних конечностей, аномалии развития лица
• Термин «синдром Клиппеля-Фейля» часто применяется у всех пациентов с врожденными аномалиями шейного отдела позвоночника, характеризующимися сращением позвонков вне зависимости от их протяженности
• Врожденное сращение шейных позвонков вследствие нарушения нормального процесса сегментации шейных сомитов (3-8 неделя эмбрионального развития)
2. Стадирование, степени и классификация аномалии Клиппеля-Фейля:
• Тип 1 (9%): массивное сращение шейных и верхне-грудных позвонков → тяжелый неврологический дефицит, часто сочетается с другими аномалиями
• Тип 2 (84%): сращение ≥ одного шейного позвоночно-двигательного сегмента
• Тип 3 (7%): сращения шейных и нижнегрудных/поясничных позвонков
3. Макроскопические и хирургические характеристики:
• Хирургические находки коррелируют с находками лучевых методов диагностики
4. Микроскопия:
• Гистологически нормальные костная ткань, межпозвонковые диски
д) Клинические особенности:
1. Клиническая картина аномалии Клиппеля-Фейля:
• Наиболее распространенные симптомы/признаки:
о Жалобы косметического характера, боль в шее или корешковая боль, медленно прогрессирующая или остро развивающаяся миелопатия:
— Массивные сращения позвонков диагностируются зачастую в младенческом/раннем детском возрасте после обнаружения соответствующих косметических дефектов
о Неврологические проблемы, возникающие в младенческом и детском возрасте, обычно являются следствием аномалий КВС
о Сращение нижних шейных позвонков (если оно не является массивным) обычно манифестирует в третьем десятилетии жизни и позднее клиникой дегенеративного поражения или нестабильности смежных с блокированными сегментов
• Другие симптомы/признаки:
о Нарушение фонации голоса (обычно при сращении > одного уровня)
о Синкинезия (зеркальные движения): 20%, верхние конечности > нижние конечности, со временем нивелируется
• Внешний вид пациента:
о Классическая триада (33-50%): короткая шея, низкое расположение границы роста волос, ограничение подвижности шейного отдела позвоночника
о Значительная вариабельность клинической и анатомической картины:
— Многие пациенты имеют абсолютно нормальный внешний вид даже несмотря на тяжесть изменений
— Наиболее постоянный клинический признак-ограничение подвижности шейного отдела позвоночника
2. Демография:
• Возраст:
о 2-3 десятилетие жизни, однако заболевание может манифестировать в любой момент на протяжении всей жизни
• Пол:
о М С3 с шейно-затылочным синостозом о Протяженный блок шейных позвонков + аномалия КВС
о Единственное нормальное межтеловое пространство между двумя блокированными сегментами
• Увеличение риска неврологического дефицита после относительно незначительной травмы вследствие гипермобильности смежных сегментов
о Пациенты высокого риска: два уровня блокированных позвонков, окципитализация атланта + базилярная инвагинация, шейный блок + стеноз позвоночника
4. Лечение:
• Исключение занятий контактными видами спорта и другими видами активности, характеризующимися высоким риском получения травм головы и шеи
• Модификация активности, иммобилизация и тракционная терапия позволяют уменьшить выраженность симптоматики
• Сохранение неврологического дефицита, выраженный болевой синдром несмотря на проводимое консервативное лечение, или прогрессирующая нестабильность → декомпрессия ± спондилодез
е) Диагностическая памятка:
1. Следует учесть:
• Большинство случаев СКФ, связанных с обращениями за медицинской помощью, и практически все случаи смерти при этом заболевании связаны с нарушением функции внутренних органов и систем
2. Советы по интерпретации изображений:
• При обследовании обращайте внимание на признаки нестабильности, прогрессирующие дегенеративные изменения, компрессию спинного мозга/ствола мозга
ж) Список использованной литературы:
1. Al-Tamimi YZ et al: Fracture through fused cervical segments following trauma in a patient with Klippel-Feil syndrome. Br J Neurosurg. 28(3):408-10, 2014
2. Jasper A et al: The multiple associations of Klippel-Feil syndrome. Acta Neurol Belg. ePub, 2014
3. Dornbos D3rd et al: Vertebral artery dissection after neck extension in an adult patient with Klippel-Feil syndrome. J Clin Neurosci. 21 (4):685—8, 2014
4. McLaughlin N et al: Klippel-Feil syndrome associated with a craniocervico-thoracic dermoid cyst. Surg Neurol Int. 4(Suppl 2): S61-6, 2013
5. Ogihara N et al: Surgical treatment of Klippel-Feil syndrome with basilar invagination. Eur Spine J. 22 Suppl 3: S380-7, 2013
6. Kumar D et al: Klippel-Feil syndrome with unilateral renal agenesis and renal failure. J Assoc Physicians India. 60:68-9, 2012
7. Apaydin M et al: Partial posterior split cervical spinal cord with Klippel-Feil syndrome. JBR-BTR. 93(1 ):30, 2010
8. Samartzis Detal: 2008 Young Investigator Award: The role of congenitally fused cervical segments upon the space available for the cord and associated symptoms in Klippel-Feil patients. Spine (Phila Pa 1976). 33( 13): 1442 — 50, 2008
9. Samartzis D et al: The extent of fusion within the congenital Klippel-Feil segment. Spine (Phila Pa 1976). 33( 1 5):1637-42, 2008
10. Smoker WR et al: Imaging the craniocervical junction. Childs Nerv Syst. 24( 1 0): 1123-45, 2008
11. Yildirim N et al: Klippel-Feil syndrome and associated ear anomalies. Am J Otolaryngol. 29(5):319-25, 2008
12. Samartzis D et al: Sprengel’s deformity in Klippel-Feil syndrome. Spine (Phila Pa 1976). 32(18): E512-6, 2007
13. Samartzis DD et al: Classification of congenitally fused cervical patterns in Klippel-Feil patients: epidemiology and role in the development of cervical spine-related symptoms. Spine (Phila Pa 1 976). 31(21): E798-804, 2006
14. Schaffer AA et al: Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome: clues from the BMP signaling pathway. Spine (Phila Pa 1976). 30( 1 2):1379-85, 2005
15. Tracy MR etal: Klippel-Feil syndrome: clinical features and current understanding of etiology. Clin Orthop Relat Res. (424): 183-90, 2004
16. Royal SA et al: Investigations into the association between cervicomedullary neuroschisis and mirror movements in patients with Klippel-Feil syndrome. AJNR Am J Neuroradiol. 23(4):724-9, 2002
17. Andronikou S et al: Klippel-Feil syndrome with cervical diastematomyelia in an 8-year-old boy. Pediatr Radiol. 31 (9):636, 2001
18. David KM et al: The dysmorphic cervical spine in Klippel-Feil syndrome: interpretations from developmental biology. Neurosurg Focus. 6(6): e1, 1999
Редактор: Искандер Милевски. Дата публикации: 18.7.2019
Синдром Клиппеля-Фейля
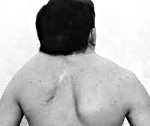
Синдром Клиппеля-Фейля — генетически детерминированная аномалия строения шейного отдела позвоночника, включающая уменьшение количества и сращение позвонков. Клинически проявляется визуально определяемым укорочением шеи, низким расположением границы роста волос на затылке, ограничением движений головой. Как правило, синдром Клиппеля-Фейля сочетается с другими врожденными аномалиями скелета и соматических органов. В диагностике участвуют различные узкие специалисты, проводится рентгенография, КТ и МРТ позвоночника, генетический анализ, расширенное обследование внутренних органов (сердца, почек, легких, головного мозга). Консервативное лечение проводится средствами массажа, ЛФК и физиотерапии. Возможно хирургическое лечение — операция цервикализации.
МКБ-10
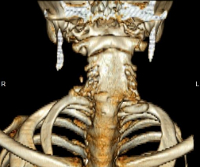
Общие сведения
Синдром Клиппеля-Фейля — врожденная, генетически обусловленная патология шейного отдела позвоночника, заключающаяся в сращении (синостозе) и уменьшении количества позвонков. Наиболее типичным и постоянным признаком синдрома является выраженное укорочение шеи, в связи с чем в медицинской практике он также упоминается как синдром короткой шеи. В большинстве случаев сочетается с другими аномалиями развития костно-мышечной системы (кривошеей, сколиозом, болезнью Шпренгеля, гипоплазией верхней конечности, синдактилией) и врожденными пороками внутренних органов (почек, сердечно-сосудистой системы, легких). Синдром Клиппеля-Фейля относится к редким заболеваниям. Частота его встречаемости — примерно 1 случай на 120 тыс. новорожденных детей. Впервые синдром был описан в 1812 г. во Франции неврологом Клиппелем и рентгенологом Фейлем, имена которых легли в основу его названия.
Современная клиническая неврология классифицирует синдром Клиппеля-Фейля на 3 типа. Первый тип — KFS1 — характеризуется уменьшенным количеством шейных позвонков. В норме у человека в шейном отделе 7 позвонков, при KFS1 обычно 4-5. Второй тип — KFS2 — синостоз всех позвонков шейного отдела, их спаянность с затылочной костью и верхнегрудными позвонками. Третий тип — KFS3 — представляет собой комбинацию первого или второго со сращением позвонков в нижнем грудном и поясничном отделах. Зачастую в шейном отделе наблюдаются добавочные ребра и spina bifida — незаращение позвонковых дужек.
Причины
Синдром Клиппеля-Фейля относится к генетически детерминированной патологии. Изредка возникают спорадические случаи синдрома. Аномалия формируется внутриутробно, еще в эмбриональном периоде за счет гипо- и аплазии, нарушения разделения шейных сегментов, запаздывания слияния в процессе закладки позвонков. Генетические аберрации гетерогенны. При KFS1 они затрагивают локус q22.1 8-й хромосомы, при KFS2 находятся в локусе q22.1 5-й хромосомы, при KFS3 — в локусе q13.31 12-й хромосомы.
Наиболее изученным является ген GDF6, ответственный за возникновение KFS1. Мутации в этом гене приводят к нарушению синтеза белка, участвующего в формировании костно-суставного аппарата путем создания разграничения между отдельными костями. В зависимости от типа синдром Клиппеля-Фейля имеет различный механизм наследования: для KFS1 и KFS3 он аутосомно-доминантный, для KFS2 — аутосомно-рецессивный.
Симптомы
Основной клинической триадой, характеризующей синдром Клиппеля-Фейля, выступает укорочение шеи, смещение границы роста волос вниз по задней поверхности шеи, нарушение подвижности позвоночника в шейном отделе. Выраженность укорочения шеи может варьировать, в наиболее тяжелом варианте мочки ушей достают плеч, а подбородок — грудины, затруднено глотание и дыхание. Характерно широкое разведение лопаток и зачастую их укорочение. Может наблюдаться типичное для болезни Шпренгеля высокое стояние одной из лопаток. В ряде случаев отмечаются аномалии мускулатуры плечевого пояса и складки на шее. В редких случаях возникает корешковый синдром — боли, связанные со сдавлением шейных спинномозговых корешков.
В 50-60% случаев синдром Клиппеля-Фейля сочетается со сколиозом, в 25% случаев — с костным вариантом кривошеи. Возможно сочетание синдрома короткой шеи с аномалиями верхних конечностей (полидактилией, синдактилией, врожденными ампутациями), деформацией стоп, пороками ребер, аномалиями зубов, асимметрией лица, дальнозоркостью. У 45% пациентов диагностируется дистопия, аплазия или гипоплазия почек, возможен гидронефроз, эктопия мочеточников. У 25% больных выявляется врожденная глухота, у 20% — волчья пасть, у 15% — врожденные пороки сердца (открытый артериальный проток, ДМЖП, ДМПП, декстрапозиция аорты). Может наблюдаться аплазия или гипоплазия легких.
Со стороны нервной системы бывает олигофрения (умственная отсталость), эпилепсия, гидроцефалия, спинно-мозговая грыжа, микроцефалия, глазодвигательные расстройства (косоглазие, птоз, синдром Горнера). С раннего возраста характерна мышечная слабость в конечностях и синкинезии — непроизвольные одновременные движения обеих рук, чаще только кистей. Со временем могут возникать спастические и вялые пара- и тетрапарезы.
Диагностика
Верификация диагноза проводится на основании наблюдаемой с рождения типичной триады признаков, данных осмотра, семейного анамнеза, результатов инструментальных и генетических исследований. Установить синдром Клиппеля-Фейля с подробным указанием имеющихся сопутствующих аномалий возможно только в результате совместной работы многих узких специалистов: невролога, ортопеда, генетика, кардиолога, нефролога, пульмонолога, офтальмолога.
В первую очередь проводится рентгенография шейного отдела позвоночника в 2-х проекциях. При KFS1 на рентгенограммах в большинстве случаев определяется полный синостоз 4-5 позвонков в единый малодифференцированный конгломерат. В ряде случаев между позвонками находятся узкие светлые полоски, соответствующие недоразвитым дискам, что говорит о частичном синостозе, который по мере роста ребенка приводит к искривлению позвоночника. Синдром Клиппеля-Фейля II типа рентгенологически характеризуется сочетанием синостозов 7 шейных позвонков с ассимиляцией атланта и сращением верхних грудных позвонков. Для исключения KFS3 проводят рентгенографию грудного и поясничного отделов позвоночника.
Более полную информацию о костных аномалиях дает КТ позвоночника. Однако ее применение в раннем детском возрасте ограниченно из-за сопутствующей исследованию лучевой нагрузки. При необходимости для оценки состояния мягкотканных структур пораженного отдела (корешков, спинного мозга) возможно проведение МРТ позвоночника. Дифференцировать синдром Клиппеля-Фейля следует от врожденной мышечной кривошеи и туберкулеза позвоночника.
Диагностический алгоритм также включает обследование состояния внутренних органов: нейросонографию, МРТ головного мозга, УЗИ брюшной полости, УЗИ сердца, ЭКГ, УЗИ или КТ почек, экскреторную урографию, рентгенографию органов грудной полости. Проводится консультирование у генетика с анализом генеалогического древа и ДНК-тестированием.
Лечение синдрома Клиппеля-Фейля
Осуществляются преимущественно консервативные лечебные мероприятия, направленные на предупреждение развития деформаций позвоночника и увеличение объема движений в шее. Проводят массаж шейного отдела и воротниковой зоны, плечевого пояса и верхних конечностей. Рекомендованы регулярные занятия лечебной физкультурой. Возможно применение физиотерапии. По показаниям проводят симптоматическое лечение нарушений в работе соматических органов. При корешковых болях назначают анальгетики, ношение воротника Шанца.
Стойкий болевой синдром, обусловленный компрессией корешков верхними ребрами, является показанием к проведению операции. Хирургическое вмешательство проводится согласно технике Бонола и представляет собой т. н. цервикализацию путем резекции верхних 4-х ребер. Доступ осуществляют через паравертебральный разрез, идущий параллельно внутреннему краю лопатки. Операция выполняется в 2-этапа, отдельно на каждой стороне.
Прогноз
Сам по себе синдром Клиппеля-Фейля имеет благоприятный витальный прогноз. Наличие пороков развития соматических органов значительно осложняет ситуацию и может выступать причиной преждевременной смерти. В функциональном отношении прогноз неблагоприятный, несмотря на проводимые консервативные мероприятия, у пациентов сохраняется выраженное ограничение движений головой, степень которого зависит от типа и тяжести синдрома. Течение заболевания может усугубиться происходящими в позвоночнике дегенеративными изменениями.