Синдром гудпасчера
Что провоцирует / Причины синдрома гудпасчера:
Этиология заболевания неизвестна. Возникновение его иногда связывают с вдыханием летучих углеводородов и других органических растворителей. Ряд авторов рассматривают синдром Гудпасчера как вариант течения идиопатического гемо- сидероза легких, что косвенно подтверждается существованием клинически и патоморфологически переходных состояний. В отличие от коллагенозов, при синдроме Гудапасчера отсутствуют явления васкулита. Предположение о вирусной природе не имеет достаточных фактических подтверждений [Spencer Н„ 1977].
Патогенез (что происходит?) во время синдрома гудпасчера:
Вопрос о первичной локализации заболевания окончательно не решен, хотя поражение легких, как правило, предшествует поражению почек.
Патологическая анатомия. При гистологическом исследовании выявляются геморрагический некротизирующий альвео- лит (изменения в легких, аналогичные идиопатическому гемо- сидерозу легких) и нефрозоиефрит с очаговыми внутрнкапил- лярными тромботическими изменениями и развитием гломеру- лярного фиброза.
Симптомы синдрома гудпасчера:
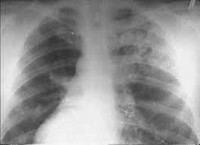
Наиболее характерными признаками синдрома Гудпасчера являются кровохарканье (повторные легочные кровотечения) и анемия. К ним часто присоединяются прогрессирующая одышка, кашель, боли в грудной клетке, похудание. На рентгенограммах выявляются мелкосетчатая деформация легочного рисунка, мелкоочаговые изменения. Может повышаться температура тела. К этим, как правило, первым признакам синдрома Гудпасчера вскоре добавляются симптомы поражения почек: протеинурия, гематурия, цилиндрурия, повышение уровня остаточного азота в крови, периферические отеки. Прогрессирует гипохромная железодефицитная анемия. При присоединении вторичной инфекции отмечаются соответствующие изменения в формуле крови.
Течение болезни неуклонно прогрессирующее и заканчивается смертью вследствие повторных легочных кровотечений или азотемической уремии.
Лечение синдрома гудпасчера:
Некоторое замедление прогрессирования процесса может дать раннее назначение кортикостерои- дов и иммуиосупрессантов (азатиоприн, циклофосфамид, 6-меркаптопурин и др.). Повторный гемодиализ несколько продлевает жизнь больным. При некупирующейся уремии (после предварительного гемодиализа) иногда производят нефрэктомию (с последующей пересадкой почек), что позволяет устранить источник антигенного раздражения. С целью удаления циркулирующих антител в последние годы применяется плазмаферез. Отмечено, что антитела против базаль- ных мембран почечных клубочков перестают определяться в сыворотке крови через 6-8 мес от начала заболевания. Поэтому интенсивная терапия в течение этого периода (кортико- стероиды, иммуносупрессанты, гемодиализ, плазмаферез, нефрэктомия), возможно, позволяет продлить жизнь больного до прекращения аутоантителогенеза.
Симптоматическая терапия включает повторные переливания крови, назначение препаратов железа.
К каким докторам следует обращаться если у Вас Синдром гудпасчера:
Синдром Гудпасчера
Синдром Гудпасчера представляет собой иммуно-воспалительное заболевание мелких сосудов легких и почек.
Симптомы Синдрома Гудпасчера:
Классической триадой, выражающей клинико-патогенетическую сущность этой болезни, являются легочные кровотечения, гломерулонефрит и антитела к антигенам основной мембраны капилляров легких и почек. Заболевание встречается редко и может поражать любой возраст, но чаще болеют молодые мужчины. Конкретные причины неизвестны; не раз описывалось развитие синдрома Гудпасчера после недавно перенесенного гриппа или вдыхания углеводородов. В связи с этим не исключено, что подобные воздействия таким образом изменяют химическую структуру упомянутых выше антигенов основных мембран, что они приобретают аутоантигенные свойства и вызывают продукцию соответствующих антител с патогенными свойствами.
Течение болезни в целом неблагоприятное, хотя и неоднотипное. На первых этапах основной угрозой являются легочные геморрагии, причем нередко больной умирает в результате первого и единственного профузного кровотечения. У ряда переживших это кровотечение может развиться относительная ремиссия легочного процесса, но во многих случаях кровохарканье и легочные геморрагии рецидивируют. Поражение почек у отдельных больных остается относительно нетяжелым, но в большинстве случаев быстро прогрессирует с развитием олигурической почечной недостаточности, приводящей к смерти в течение нескольких месяцев и даже недель. Средняя продолжительность жизни у неадекватно леченных больных составляет менее полугода.
Диагностика Синдрома Гудпасчера:
При дифференциальном диагнозе синдрома Гудпасчера следует иметь в виду, что сочетание легочного кровотечения с выраженной почечной патологией (в том числе с недостаточностью почек) может встретиться при гранулематозе Вегенера, узелковом полиартериите, СКВ, геморрагическом васкулите, криоглобулинемии, эмболии ветвей легочной артерии вследствие тромбоза почечной вены, болезни легионеров и даже при тяжелой недостаточности кровообращения с выраженным застоем в легких и почках. Однако все эти заболевания отличаются от синдрома Гудпасчера по клиническим признакам и отсутствию антител к антигенам базальных мембран.
Лечение Синдрома Гудпасчера:
К каким докторам следует обращаться если у Вас Синдром Гудпасчера:
Синдром Гудпасчера
Синдром Гудпасчера – аутоиммунная патология, характеризующаяся образованием аутоантител к базальным мембранам почечных клубочков и легочных альвеол. Клинически синдром Гудпасчера проявляется рецидивирующими легочными кровотечениями, прогрессирующим гломерулонефритом и почечной недостаточностью. Диагноз синдрома Гудпасчера подтверждается выявлением антител к базальной мембране клубочков (Anti GBM), данными биопсии почек и легких, рентгенологического исследования легких. Лечение синдрома Гудпасчера включает иммуносупрессивную терапию (глюкокортикостероиды, цитостатики), плазмаферез, по показаниям – гемодиализ, трансплантацию почек.
Общие сведения
Причины синдрома Гудпасчера
Этиологические механизмы заболевания достоверно не определены. Клинические наблюдения указывают на связь развития синдрома Гудпасчера с перенесенной вирусной инфекцией (гриппом, вирусным гепатитом А и др.), приемом лекарственных препаратов (карбимазола, пеницилламина), производственными вредностями (вдыханием паров органических растворителей, лаков, бензина), переохлаждением, курением. Отмечена генетическая предрасположенность к данному синдрому у лиц–носителей HLA-DRwl5, HLA-DR4 и HLA-DRB1 аллелей. Описаны семейные случаи синдрома Гудпасчера.
Под воздействием того или иного этиологического фактора, в результате изменения толерантности иммунной системы, в организме начинается выработка аутоантител к базальным мембранам легочных альвеол и почечных клубочков. Предполагается, что в роли аутоантигена выступает структурный компонент а-3 цепи коллагена IV типа, присутствующий в базальных мембранах легочных и почечных капилляров. Сформировавшиеся антитела (GВМ-антитела) в присутствии С3-комплемента связываются с антигенами; образовавшиеся иммунные комплексы откладываются вдоль базальных мембран, индуцируя иммуно-воспалительное поражение почечных клубочков (гломерулонефрит) и альвеол (альвеолит). В развитии аутоиммунного воспаления большая роль принадлежит активации клеточных элементов (Т-лимфоцитов, эндотелиоцитов, моноцитов, альвеолярных макрофагов, полиморфноядерных лейкоцитов), цитокинов (инсулиноподобного, тромбоцитарного факторов роста, факторов некроза опухоли, интерлейкина-1), свободных радикалов, протеолитических ферментов и других факторов, повреждающих почечную и легочную ткань.
Патоморфологическими субстратами синдрома Гудпасчера служат геморрагический некротизирующий альвеолит и нефрозонефрит. При гистологическом исследовании почечной ткани обнаруживается пролиферативно-мембранозный, пролиферативный или некротизирующий гломерулонефрит, склероз клубочков и фиброз почечной паренхимы. Морфологическое исследование легочной ткани выявляет капиллярит межальвеолярных перегородок, легочные инфильтраты, гемосидероз, пневмосклероз.
Симптомы синдрома Гудпасчера
Выделяют три варианта клинического течения синдрома Гудпасчера: злокачественный, умеренный и медленный. Для злокачественного течения характерны рецидивирующая геморрагическая пневмония и стремительно прогрессирующий гломерулонефрит. При втором типе легочно-почечный синдром развивается медленнее и выражен умеренно. При третьем варианте синдрома Гудпасчера преобладают явления гломерулонефрита и ХПН; легочные проявления развиваются поздно.
Вскоре к легочным проявлениям добавляется почечная симптоматика: гематурия, олигурия, периферические отеки, артериальная гипертензия. У 10-15% пациентов синдром Гудпасчера манифестирует с клинических признаков гломерулонефрита. Во многих случаях течение заболевания сопровождается миалгиями, артралгиями, геморрагиями кожи и слизистых оболочек, интраретинальными кровоизлияниями, перикардитами.
Диагностика синдрома Гудпасчера
При осмотре пациентов с синдромом Гудпасчера обращает на себя внимание бледность кожных покровов, пастозность или отеки лица. В легких выслушиваются сухие и влажные хрипы, количество которых увеличивается в момент кровохарканья и после него. В общем анализе крови обнаруживается гипохромная анемия, анизоцитоз, пойкилоцитоз, лейкоцитоз, резкое увеличение СОЭ. Для общего анализа мочи характерна протеинурия, цилиндрурия, эритроцитурия; проба по Зимницкому выявляет изогипостенурию. В биохимическом анализе крови определяется нарастание уровня креатинина, мочевины, серомукоида; снижение концентрации железа. Для синдрома Гудпасчера типично обнаружение большого количества эритроцитов, сидерофагов и гемосидерина в общем анализе мокроты.
Наиболее чувствительным и специфичным методом диагностики синдрома Гудпасчера служит определение антител к базальной мембране клубочков (Anti-GBM) с помощью ИФА или РИА. На рентгенограммах легких выявляются множественные очаговые тени. Морфологическое подтверждение синдрома Гудпасчера основывается на данных биопсии легких и почек. Вспомогательное значение имеют результаты инструментальной диагностики: спирометрии, УЗИ почек, ЭКГ, ЭхоКГ.
Лечение и прогноз синдрома Гудпасчера
В остром периоде синдрома Гудпасчера показано назначение пульс-терапии метилпреднизолоном или комбинированной пульс-терапии (метилпреднизолон+циклофосфан) с последующим переводом на поддерживающую терапию после достижения клинико-лабораторной и рентгенологической ремиссии. С целью элиминации циркулирующих иммунных комплексов проводится плазмаферез. Симптоматическая терапия синдрома Гудпасчера включает переливания эритроцитарной массы и плазмы крови, назначение препаратов железа. При развитии терминальной почечной недостаточности применяются сеансы гемодиализа. Возможно выполнение нефрэктомии с последующей трансплантацией почки, однако это не исключает рецидива некротизирующего гломерулонефрита в трансплантате.
Течение синдрома Гудпасчера неуклонно прогрессирующее; прогноз – мало обнадеживающий. Гибель пациентов происходит, как правило, вследствие профузных легочных кровотечений, тяжелой почечной или дыхательной недостаточности. При злокачественном варианте летальный исход наступает в считанные недели; в остальных случаях средняя продолжительность жизни колеблется от нескольких месяцев до 1-3 лет. В литературе описаны единичные спонтанные ремиссии синдрома Гудпасчера.
Синдром Гудпасчера
 Синдром Гудпасчера — заболевание, характеризующееся сочетанием патологических изменений в легких (лёгочный гемосидероз) и почках (гломерулонефрит).
Синдром Гудпасчера — заболевание, характеризующееся сочетанием патологических изменений в легких (лёгочный гемосидероз) и почках (гломерулонефрит).
Синонимы:
Впервые описан Goodpasture в 1919 г. Синдром Гудпасчера — редкое заболевание аутоиммунной природы, при котором в патологический процесс вовлекаются базальные мембраны сосудов лёгких и почек. Болеют преимущественно мужчины в возрасте 18-35 лет. В качестве пускового агента рассматривается вирусная инфекция (косвенным подтверждением является увеличение частоты заболевания в период эпидемий гриппа). В основе патогенеза Синдрома Гудпасчера лежит цитотоксическая тканевая реакция. Под влиянием неустановленных факторов формируется аутоиммунная реакция с поражением базальных мембран альвеол, мелких сосудов лёгких и почек по типу васкулита (воспаления стенки мелких сосудов). Скорее всего, у генетически предрасположенных людей, под влиянием вирусных инфекций, курения и других неблагоприятных факторов, стимулируется продукция антител, возникает заболевание или его обострение.
Основные клинические симптомы
Лихорадка. Резкая слабость, кровохарканье (лёгочное кровотечение). Больной во время кашля выделяет пенистую ярко-красную мокроту. Одышка. Непостоянные боли в груди. Параллельно отмечается симптоматика гломерулонефрита: мочевой синдром (низкий удельный вес мочи, протеинурия, гематурия), отёки. При прогрессировании поражения почек развивается картина почечной недостаточности. Появление отёков, сонливости, неукротимой рвоты, анурии свидетельствует о развитии уремии. Повышение артериального давления отмечается у 15 % больных, гепатоспленомегалия — у 10%.
Дифференциальная диагностика основана на исключении заболеваний, сопровождающихся кровохарканьем:
Прогноз неблагоприятный. Средняя продолжительность жизни составляет 8-10 мес. Причинами летального исхода являются лёгочное кровотечение или прогрессирующая почечная недостаточность.
Публикации в СМИ
Синдром Гудпасчера
Этиология неизвестна. К факторам риска относят: курение, респираторную инфекцию, контакт с летучими углеводородами.
Генетические аспекты. Выявлена ассоциация с HLA DRw2.
Патоморфология • Почки •• Эпителиальноклеточные полулуния •• Сморщивание почечных клубочков •• Интерстициальный воспалительный экссудат • Лёгкие •• Внутриальвеолярные кровоизлияния •• Макрофаги, нагруженные гемосидерином •• Фиброз межальвеолярных перегородок.
Клиническая картина • Поражение лёгких (опережает поражение почек на несколько недель или месяцев); варьирует от небольшой одышки до повторных лёгочных кровотечений с развитием дыхательной недостаточности • Быстропрогрессирующий гломерулонефрит с развитием почечной недостаточности • Артериальная гипертензия (20%) • Гриппоподобный синдром: лихорадка, миалгии, артралгии, слабость.
Лабораторные данные • ОАК •• гипохромная ЖДА • ОАМ •• микрогематурия •• протеинурия, не достигающая нефротического порога • АТ против Аг базальной мембраны клубочков почек (радиоиммунный метод или метод непрямой иммунофлюоресценции) • Перинуклеарные антинейтрофильные АТ.
Инструментальные данные • Биопсия почек — отложения иммуноглобулинов и комплемента в базальной мембране клубочков, экстракапиллярные полулуния в 50% клубочков • Рентгенография органов грудной клетки — летучие асимметричные облаковидные инфильтраты; деструкция лёгких не характерна.
Дифференциальная диагностика • Гранулематоз Вегенера протекает с деструкцией лёгочной ткани и поражением верхних дыхательных путей • Микроскопический полиангиит, помимо лёгочного и почечного синдромов, имеет и другие (множественный мононеврит, кожные изменения) • Тромбоз почечных вен и ТЭЛА (острое развитие, отсутствие иммунологической активности) • Быстропрогрессирующий гломерулонефрит, осложнённый отёком лёгких.
ЛЕЧЕНИЕ
Общая тактика предполагает сочетание плазмафереза, активной иммунодепрессивной терапии и методов коррекции почечной функции. Диета соответствует диете при почечной недостаточности.
Лекарственное лечение • ГК •• Пульс-терапия метилпреднизолоном 30 мг/кг/сут в/в капельно в течение 20–30 мин 3 дня подряд •• Преднизолон после пульс-терапии в дозе 2 мг/кг внутрь до клинического эффекта с постепенным снижением дозы: до 1,75 мг/кг в течение 1 мес, 1,5 мг/кг в течение 3 мес, далее каждую дозу назначают по 6 мес (1,25–1,0–0,75–0,5–0,25 мг/кг), далее каждую дозу назначают по 12 мес (0,125–0,0625 мг/кг). Больным старше 60 лет дозу следует снизить на 25%.
• Цитотоксические иммунодепрессанты применяют в комбинации с ГК •• Циклофосфамид 2–3 мг/кг/сут •• Азатиоприн 1–2мг/кг/сут. Примечания: дозу препарата следует уменьшить на 50% при снижении СКФ до 10 мл/мин; необходим контроль за количеством лейкоцитов и тромбоцитов крови.
Немедикаментозная терапия • Плазмаферез ежедневно или через день в сочетании с иммунодепрессивной терапии в течение 1–2 нед • Гемодиализ — при развитии почечной недостаточности.
Хирургическое лечение • Трансплантация почки в терминальной стадии ХПН. Рецидива заболевания в трансплантате не наблюдают, если перед операцией в крови не обнаруживались АТ к базальной мембране клубочков.
Прогноз неблагоприятен, если на момент постановки диагноза (до начала лечения) уже имеются признаки почечной недостаточности.
Синоним. Синдром лёгочно-почечный наследственный.
МКБ-10 • M31.0 Гиперчувствительный ангиит
Код вставки на сайт
Синдром Гудпасчера
Этиология неизвестна. К факторам риска относят: курение, респираторную инфекцию, контакт с летучими углеводородами.
Генетические аспекты. Выявлена ассоциация с HLA DRw2.
Патоморфология • Почки •• Эпителиальноклеточные полулуния •• Сморщивание почечных клубочков •• Интерстициальный воспалительный экссудат • Лёгкие •• Внутриальвеолярные кровоизлияния •• Макрофаги, нагруженные гемосидерином •• Фиброз межальвеолярных перегородок.
Клиническая картина • Поражение лёгких (опережает поражение почек на несколько недель или месяцев); варьирует от небольшой одышки до повторных лёгочных кровотечений с развитием дыхательной недостаточности • Быстропрогрессирующий гломерулонефрит с развитием почечной недостаточности • Артериальная гипертензия (20%) • Гриппоподобный синдром: лихорадка, миалгии, артралгии, слабость.
Лабораторные данные • ОАК •• гипохромная ЖДА • ОАМ •• микрогематурия •• протеинурия, не достигающая нефротического порога • АТ против Аг базальной мембраны клубочков почек (радиоиммунный метод или метод непрямой иммунофлюоресценции) • Перинуклеарные антинейтрофильные АТ.
Инструментальные данные • Биопсия почек — отложения иммуноглобулинов и комплемента в базальной мембране клубочков, экстракапиллярные полулуния в 50% клубочков • Рентгенография органов грудной клетки — летучие асимметричные облаковидные инфильтраты; деструкция лёгких не характерна.
Дифференциальная диагностика • Гранулематоз Вегенера протекает с деструкцией лёгочной ткани и поражением верхних дыхательных путей • Микроскопический полиангиит, помимо лёгочного и почечного синдромов, имеет и другие (множественный мононеврит, кожные изменения) • Тромбоз почечных вен и ТЭЛА (острое развитие, отсутствие иммунологической активности) • Быстропрогрессирующий гломерулонефрит, осложнённый отёком лёгких.
ЛЕЧЕНИЕ
Общая тактика предполагает сочетание плазмафереза, активной иммунодепрессивной терапии и методов коррекции почечной функции. Диета соответствует диете при почечной недостаточности.
Лекарственное лечение • ГК •• Пульс-терапия метилпреднизолоном 30 мг/кг/сут в/в капельно в течение 20–30 мин 3 дня подряд •• Преднизолон после пульс-терапии в дозе 2 мг/кг внутрь до клинического эффекта с постепенным снижением дозы: до 1,75 мг/кг в течение 1 мес, 1,5 мг/кг в течение 3 мес, далее каждую дозу назначают по 6 мес (1,25–1,0–0,75–0,5–0,25 мг/кг), далее каждую дозу назначают по 12 мес (0,125–0,0625 мг/кг). Больным старше 60 лет дозу следует снизить на 25%.
• Цитотоксические иммунодепрессанты применяют в комбинации с ГК •• Циклофосфамид 2–3 мг/кг/сут •• Азатиоприн 1–2мг/кг/сут. Примечания: дозу препарата следует уменьшить на 50% при снижении СКФ до 10 мл/мин; необходим контроль за количеством лейкоцитов и тромбоцитов крови.
Немедикаментозная терапия • Плазмаферез ежедневно или через день в сочетании с иммунодепрессивной терапии в течение 1–2 нед • Гемодиализ — при развитии почечной недостаточности.
Хирургическое лечение • Трансплантация почки в терминальной стадии ХПН. Рецидива заболевания в трансплантате не наблюдают, если перед операцией в крови не обнаруживались АТ к базальной мембране клубочков.
Прогноз неблагоприятен, если на момент постановки диагноза (до начала лечения) уже имеются признаки почечной недостаточности.
Синоним. Синдром лёгочно-почечный наследственный.
МКБ-10 • M31.0 Гиперчувствительный ангиит