Синдром гольденхара фото что это такое
Синонимы синдрома Гольденхара. Глазо-ушной синдром. Глазо-ушная дисплазия. Окуло-аурикуло-вертебральная дисплазия.
Определение синдрома Гольденхара. Характерный комбинированный порок развития глазо-ушной области в результате задержки развития и дифференциации в районе первой жаберной дуги или первой жаберной борозды. Относится к синдромам дисморфии черепно-нижнечелюстно-лицевой, синдромам дуги нижней челюсти, глазо-зубным синдромам.
Автор. Goldenhar Maurice — разрабатывал эту проблему в своей докторской диссертации в Университетской глазной клинике в Женеве (1952). Он же опубликовал первые наблюдения синдрома.
Симптоматология синдрома Гольденхара:
1. Эпибульбарный дермоид, или липодермоид, или субконъюнктивальная липома.
2. Аурикулярный или преаурикулярный придаток.
3. Преаурикулярная фистула.
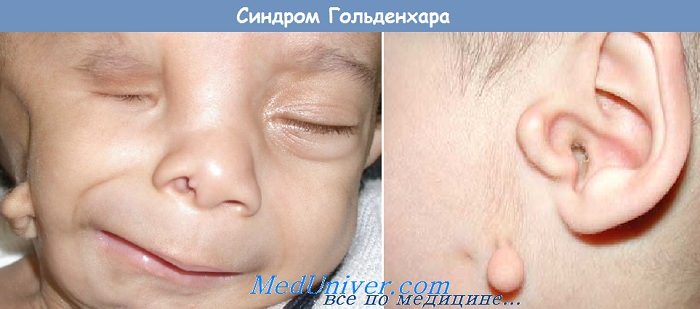
4. Пороки развития ушной раковины (аплазия, дисплазия, асимметрия). Может наблюдаться также атрезия наружного слухового прохода.
5. Гипоплазия половины лица.
6. Макростомия.
7. Аномалии расположения зубов.
Этиология и патогенез синдрома Гольденхара. Нозологическое место синдрома не ясно. См.также S. Franceschetti I, к которому он весьма близок.
Дифференциальный диагноз. Другие пороки развития из числа синдромов черепно-нижнечелюстно-лицевой дисморфии, особенно S. Franceschetti I (см.) и S. Wildervanck (см.). Отовертебральный синдром. См. также синдромы нижней челюсти и глазо-зубной синдром.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром гольденхара фото что это такое
Гемифациальная микросомия является частью окуло-аурикуло-вертебрального синдрома. Наиболее часто встречающейся клинической формой является синдром Гольденхара. Основными признаками его являются пороки развития ушной раковины, которая обычно расположена на половине лица с микросомией, эпибульбарные дермоиды, пороки развития позвоночника, колобома верхнего века, преаурикулярная папиллома, агенезия околоушной слюнной железы с одной стороны, врожденный паралич лицевого нерва.
Выраженность микросомии нижней челюсти коррелирует с общей тяжестью заболевания. Примерная частота встречаемости составляет 1:5600, причина неизвестна. Мужчины и женщины подвержены в равной степени. У трети пациентов аномалии развиваются на обеих половинах лица, у остальных 2/3 — только на одной. Правая и левая сторона поражаются одинаково часто. Окуло-аурико-вертебральная диспалазия является генетически гетерогенным синдромом. И хотя некоторые случаи синдрома Гольденхара были вызваны делециями в хромосомах 5р15 и 14q32, схожие фенотипы наблюдаются у пациентов с трисомиями 18,7,9 хромосом, а также терминальной делецией 22q.
И хотя в большинстве случаев заболевание развивается спонтанно, генеалогические исследования показали, что возможен как аутосомно-рецессивный, так и аутосомно-доминантный тип наследования. С аналогичным фенотипом связаны и некоторые факторы со стороны матери: сахарный диабет, прием ретиноевой кислоты, талидомида, употребление кокаина. Поскольку поражаются обычно производные первые и второй жаберной дуги, к развитию классической клинической картины часто приводит разрыв стременной артерии во время внутриутробного развития. Схожесть проявлений окуло-аурикуло-вертебральной дисплазии и CHARGE-синдрома: (С [coloboma]—колобома глаз; Н [heart]—пороки сердца; A [atresia]—атрезия хоан, R [retarded growth] — задержка роста и развития; G [genital] — пороки развития мочеполовой системы, Е [ear] — глухота и пороки развития уха) позволяют предположить, что в основе патофизиологии синдрома лежит нарушение развития клеток нервного гребня.
У детей с гемифациальной микросомией более чем в половине случаев имеются пороки других органов и систем (скелета, сердца, легких, центральной нервной системы, желудочно-кишечного тракта), причем их выраженность коррелирует с проявлениями микросомии. Нарушения формирования экстракраниального скелета имеются у 40-60% пациентов, наиболее часто поражаются позвоночный столб и ребра. У 25-50% детей с гемифациальной микросомией также имеются пороки сердца (наиболее часто —дефект межжелудочковой перегородки).
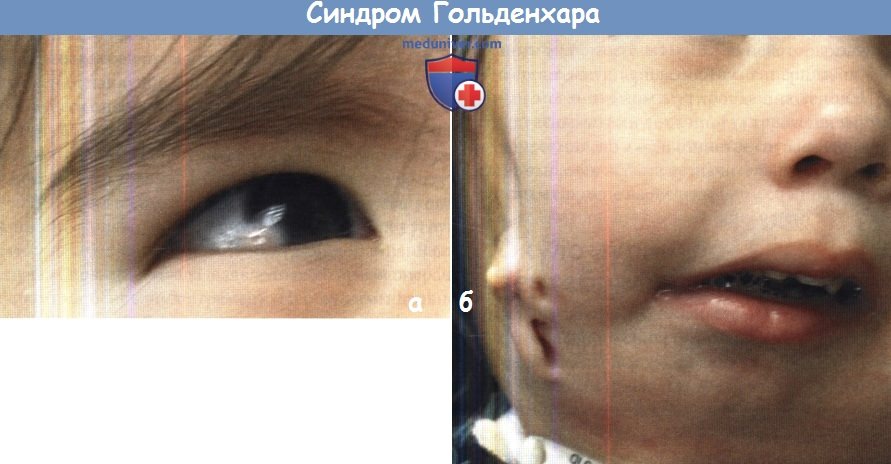 (а) Эпибульбарный дермоид у пациента с синдромом Гольденхара.
(а) Эпибульбарный дермоид у пациента с синдромом Гольденхара.
(б) Для синдрома Гольденхара характерны микротия, аномальный ход лицевого нерва,
гемифациальная и гемимандибулярная микросомия.
У 70-75% пациентов имеется кондуктивная или смешанная тугоухость. Возможно наличие микротии, атрезии наружного слухового прохода, мальформация или слияние слуховых косточек, отсутствие овального окна. Повышен риск воспалительных заболеваний среднего уха, часто эффективной оказывается установка тимпа-ностомических трубок на длительное время. В редких случаях встречается дисплазия улитки и полукружных каналов, ведущая к нейросенсорной тугоухости. Пациентам с окуло-аурикуло-вертебральным синдромом необходимо выполнять КТ височных костей перед любой операцией на среднем ухе, т.к. у них часто встречаются аномалии расположения лицевого нерва.
Результаты хирургического лечения по поводу микротии или атрезии слухового прохода у детей с окуло-аурикуло-вертебральным синдромом обычно хуже, чем в изолированных случаях; это объясняется сочетанными нарушениями анатомии и роста костей лицевого скелета.
Речь обычно развивается нормально, т.к. слух на одном ухе чаще всего сохранен. У пациентов с двусторонним снижением слуха используются традиционные слуховые аппараты или аппараты с проведением по кости. При выраженной двусторонней нейросенсорной тугоухости возможно проведение кохлеарной имплантации. Ключевым моментом лечения таких больных являются занятия с логопедом, т.к. слабость половины языка может вызывать нарушения артикуляции. Нарушения координации языка также могут вызывать проблемы с питанием.
Все новорожденные, у которых во время кормления возникает кашель или поперхивания, должны обследоваться на предмет трахеопищевой фистулы, т.к. у данной группы пациентов отмечается высокая частота встречаемости.
Краниофациальная реконструкция показана только в тяжелых случаях, обычно она ограничивается проведением дистракционного остеогенеза нижней челюсти. Также имеются сообщения о реконструкции верхней челюсти по Le Fort I и реконструкция нижней челюсти свободным лоскутом. Сведения о результатах зачастую разнятся. Во многих случаях для достижения приемлемого функционального и косметического результата требуется проведение нескольких оперативных вмешательств.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Гольденхара
Медицинский эксперт статьи


Окуло-аурикуло-вертебральная дисплазия, как еще называется эта достаточно редкая врожденная патология, обычно затрагивает развитие органов одной половины лица: глаз, уха, носа, мягкого неба, губ, челюсти. Она является отдельным видом лицевой микросомии, при которой из-за внутриутробного недоразвития скелетных, нервно-мышечных и прочих компонентов мягких тканей (производных I и II жаберных щелей) наружные органы одной половины лица отличаются заметно более мелкими размерами. Очень редко эта патология бывает двусторонней.
 [1], [2], [3], [4], [5]
[1], [2], [3], [4], [5]
Код по МКБ-10
Эпидемиология
Медицинская статистика свидетельствует, что заболевания окуло-аурикуло-вертебрального спектра в структуре внутриутробных аномалий черепно-лицевой зоны следует за такими патологиями как заячья губа и волчья пасть, как сочетанных, так и отдельных. Частота появления синдрома Гольденхара, упоминаемая в зарубежной медицинской литературе, – один ребенок из 3,5-7 тысяч рожденных живыми младенцев. Этот синдром диагностируется у одного из тысячи глухих новорожденных. Примерно в 70% случаев поражения носят односторонний характер, при двусторонних дефектах они более выражены с одной стороны, причем среди них превалирует правая сторона с частотой встречаемости 3:2. Распределение по половому признаку – на троих детей мужского пола припадает две девочки.
[6], [7], [8], [9]
Причины синдрома Гольденхара
Сочетание дисплазии глаз, ушей и позвоночника, описанное в начале 50-х годов прошлого века американским доктором М.Гольденхаром, увековечилось его именем. Это редкая врожденная патология пока еще мало изучена, но мнение большинства исследователей совпадает в том, что этиологически он обусловлен генетической предрасположенностью. Тип наследования в настоящее время не определен, случаи заболевания носят спорадический характер. Есть сообщения об аутосомно-доминантном семейном наследовании. У пациентов с этим заболеванием выявляются хромосомные аномалии. Гипотетически факторы риска рождения ребенка с диспластическим поражениями окуло-аурикуло-вертебральных структур – это и кровнородственный брак, и предшествующие рождению ребенка аборты у матери, и тератогенез экзогенной или эндогенной природы, в частности, сахарный диабет или алиментарное ожирение будущей мамы.
Степень риска рождения больного ребенка у генетического носителя составляет 3%, а повторение рождения ребенка с этим дефектом в одной семье приравнивается к 1%.
[10], [11]
Патогенез
Патогенез, опять же, гипотетически, основан на возможности кровоизлияния на участке I и II жаберных щелей эмбриона, совпадающего по времени с заменой источника кровоснабжения в данной зоне. Подача крови из стременной артерии замещается снабжением из наружной сонной артерии. Сосудистый инсульт, произошедший в данное время и в данном месте, приводит к патологическим трансформациям клеточной пролиферации и аномальному формированию костно-мышечных, нервных и прочих элементов мягких тканей, развивающихся из производных I и II жаберных щелей.
[12], [13], [14], [15], [16], [17], [18], [19], [20]
Симптомы синдрома Гольденхара
Первые признаки наличия лицевой микросомии в основном определяются визуально при осмотре новорожденного. Типичными симптомами являются некоторая ассиметрия лица, нарушение размеров и положения глазницы, деформация ушных раковин в форме специфических аурикулярных «выступов», при этом другие изменения внешнего уха могут отсутствовать, недоразвитие нижней челюсти.
С ростом ребенка симптоматика становится все более заметной. Фенотип Голденхара включает аномалии развития уха (микротию), глаз, носа, мягкого неба, губ и челюсти. Одним из типичных симптомов считается наличие хористом (эпибульбарных дермоидов) на поверхности глазного яблока. Это опухолевые образования, содержащие не характерные для их локализации ткани (волосяные фолликулы, сальные и потовые железы, фиброжировую ткань). Данный симптом специфичен для 70% случаев синдрома Гольденхара. Из офтальмологических мальформаций могут присутствовать (25% случаев и более) – липодермоиды в наружной области конъюнктивы глазного яблока, коломбы верхнего века, пороки глазодвигательной мускулатуры, разрез глаз с опущенными вниз внешними уголками глазных яблок. Изредка (не чаще 5% случаев) наблюдается малый диаметр роговицы глаза, сквозной дефект или отсутствие радужной оболочки, опущение верхнего века, недоразвитие глазного яблока и его малые размеры, косоглазие и катаракта.
Аномалии развития ушных раковин встречаются чаще всего. Они деформированы и заметно меньше нормы по размеру (примерно 80% пациентов), у половины пациентов с синдромом имеют аномальное расположение, может отсутствовать наружный слуховой проход (40% пациентов). У 55 % больных наблюдались дефекты развития среднего уха и отсутствие слуха.
Очень характерный признак синдрома Гольденарха (85%) –недоразвитие отростков нижней челюсти, также достаточно часто асимметричны и недоразвиты лицевые мышцы, верхняя и нижняя челюсть. При осмотре ротовой полости наблюдается небо в форме высокой арки, иногда с расщелиной, открытый прикус, слишком широкая ротовая щель, расщелина язычка и добавочные уздечки.
Чуть меньше половине случаев сопутствовали недоразвития позвонков, чаще шейного отдела – клиновидные, слитые, полупозвонки, сколиоз, трети – spina bifida, пороки развития ребер, пятой части – косолапость.
Менее трети случаев синдрома Гольденхара сопровождались аномалиями развития сердечно-сосудистой системы (пороком межжелудочковой перегородки, открытым Боталловым протоком, тетрадой Фалло, сужением либо полным перекрытием аорты). Умственная отсталость в разной степени наблюдалась у десятой части пациентов с этим синдромом.
Существует несколько классификаций, отражающих стадии заболевания, а точнее степени его тяжести. Наиболее полная – OMENS. Она выделяет три стадии выраженности поражения каждого из объектов мальформаций при гемифациальной микросомии: глаза (orbit), нижняя челюсть (mandible), ухо (ear), лицевой нерв (facial nerve) и кости скелета (skeletal). Поскольку дефекты множественные и каждая структура обычно поражена в разной степени, то выглядит это примерно так: O2M3E3N2S1*. Звездочка отражает наличие дополнительных пороков нечерепно-лицевых объектов.
Классификация SAT обращает внимание на три основных объекта: скелет (skeletal), раковина уха (auricle), мягкие ткани (soft tissue). Согласно данной классификации пороки развития скелета рассматриваются по пяти стадиям (от S1 до S5), нарушения структуры ушной раковины – по четырем (от АО до A3); дефекты мягких тканей – по трем (от Т1 до Т3). Так, самая легкая стадия заболевания – S1A0T1, тяжелые пороки развития – S5A3T3. Система SAT проигрывает в сравнении с предыдущей по отсутствию важных объектов поражения, которые в ней не отражаются.
Некоторые авторы выделяют виды гемифациальной микросомии по фенотипу, связанному с объектами поражений. В этой классификации тип Гольденхара выделен в отдельный вид с присущими ему специфическими пороками развития.
[21], [22], [23], [24]
Осложнения и последствия
Последствия и осложнения этой врожденной патологии находятся в прямой зависимости от степени тяжести пороков развития, некоторые из которых несовместимы с жизнью, другие же, например, неправильный прикус – могут вызвать ряд неудобств. Очень много зависит от своевременно начатого лечения. Если время упущено, то у ребенка с данной патологией проявляется гипоплазия лицевых костей, причем прогрессирующая и все более заметная. Становится трудно совершать глотательные и жевательные движения. Патологии зрения, слуха тоже будут прогрессировать. Итогом всех ухудшений будут серьезные физические неудобства и психологический дискомфорт, что скажется на качестве жизни ребенка и его родителей.
[25], [26], [27], [28], [29], [30]
Диагностика синдрома Гольденхара
Как правило, предварительный диагноз этой врожденной аномалии устанавливается уже у новорожденного при выявлении асимметрии лица в сочетании с другими специфическими визуальными симптомами.
Для уточнения диагноза данного заболевания используются разнообразные диагностические процедуры. Одна из первых – определение остроты слуха, поскольку поражения наружного и внутреннего уха встречаются почти в каждом случае и в первую очередь обращают на себя внимание. Раннее исследование слуха вызвано необходимостью профилактики отставания ребенка в психоречевом развитии. В раннем возрасте ребенка диагностируют во время сна. Используются такие методы: импедансометрия, регистрация слуховых вызванных потенциалов (электрокохлеография, отоакустическая эмиссия), компьютерная аудиометрия.
Детей постарше тестируют в игровой форме с помощью речевой аудиометрии. Инструментальную и субьективную диагностику слуха показано проводить каждые шесть месяцев на протяжении семи лет.
Первоочередные консультации должны быть у челюстно-лицевого хирурга, офтальмолога, ортодонта, ортопеда, отоларинголога, чтобы диагностировать максимально возможные пороки развития. Инструментальную диагностику и анализы специалисты назначают по необходимости, в зависимости от выявленных патологий. Обычно назначают электрокардиографию, ренгенографию, ультразвуковое исследование внутренних органов.
Ребенку после достижения трехлетнего возраста назначается компьютерная томография височных зон.
Дети, имеющие этот диагноз, требуют консультаций многих специалистов в зависимости от наличия пороков развития: сурдолога, логопеда-дефектолога, кардиолога, нефролога, невропатолога и прочих.
[31], [32], [33]
Какие анализы необходимы?
Дифференциальная диагностика
Дифференциальная диагностика проводится с другими врожденными черепно-лицевыми пороками развития, такими как: дизостозы – нижнечелюстно-лицевой, гемифациальный, акрофациальный, другие виды гемифациальной микросомии, синдромы – Кауфмана и рото-лице-пальцевой, ассоциация Чарджа.
К кому обратиться?
Лечение синдрома Гольденхара
Многообразие пороков развития черепа и позвоночника, а также других органов и систем у пациентов с данной врожденной патологией приводит к многоэтапному лечению у многих специалистов. При легких степенях поражения ребенка наблюдают до трех лет, а затем начинается хирургическое лечение.
В случаях тяжелых врожденных дефектов сначала применяется оперативное лечение (в младенчестве или до достижения двух лет). После этого проводят симптоматическое комплексное лечение. Синдром Гольденхара без многоэтапного хирургического вмешательства вылечить нельзя. Количество и объем хирургических операций зависят от степени тяжести патологий. Таким больным проводят обычно компрессионно-дистракционный остеосинтез; эндопротезирование височно-нижнечелюстного сустава, нижней и верхней челюсти; остеотомию носа, нижней и верхней челюстей, исправляющую пороки их развития и патологический прикус; пластические операции (гениопластика, ринопластика). Для профилактики воспалительных осложнений и ускорения процесса реабилитации назначается антибиотикотерапия и витаминотерапия. При челюстно-лицевых хирургических манипуляциях назначаются, как правило, остеотропные антибиотики: пенициллины, линкомицин, эритромицин.
Пенициллины – естественные соединения, синтезируемые разными формами пенициллинового плесневого гриба и полусинтетические, на основе 6-аминопенициллановой кислоты, выделенной из естественных соединений. Их антибактериальная способность основана на нарушении клеточной оболочки бациллы. Они малотоксичны, имеют широкий спектр дозировок, однако, большинство лекарственных аллергий вызвано именно антибиотиками пенициллинового ряда.
Линкомицин – антибиотик выбора при аллергии к пенициллинам, может назначаться детям с месячного возраста, терапевтические дозировки препарата оказывают бактериостатический эффект, более высокие – бактерицидный, применяется для устранения инфицирования костей, суставов, мягких тканей. Противопоказан при тяжелых нарушениях почечной и печеночной функции. Может вызывать аллергию.
Эритромицин – относится к группе макролидных анибактериальных средств, имеет широкий спектр бактерицидного действия, может применяться в офтальмологии. Детям назначается с года. Одно из побочных действий этого препарата, а также реакция на передозировку – нарушение слуха, однако, оно считается обратимым. Поэтому препарат может быть назначен при непереносимости двух предыдущих, тем более, что профилактическая антибиотикотерапия назначается кратковременно. Ее цель – достичь к моменту операции наибольшей терапевтической плотности препарата в тканях. Профилактическое лечение начинают за час или два перед операцией и прекращают через двое-трое суток.
В зависимости от наличия болевых ощущений назначаются анальгетики. Маленьким пациентам назначают детский Нурофен, обладающий быстродействием, а также обеспечивающий жаропонижающий и противовоспалительный эффект и достаточно продолжительное действие (до восьми часов). Максимальная дозировка не должна превышать 30мг на килограмм веса ребенка ежесуточно.
В реабилитационный период нужно обеспечить ребенку полноценное питание и витамины. Назначаются поливитаминные комплексы, включающие аскорбиновую кислоту, ретинол, токоферол, витамины группы D и B.
Для предупреждения инфицирования и рассасывания послеоперационных отеков и инфильтратов применяют физиотерапевтическое лечение ультрафиолетовым излучением, ультразвуковыми и электромагнитными волнами, а также – лазерную и магнитную терапию и их сочетание, гипербарическую оксигенацию.
Ортодонтическое лечение предусматривают профилактику ассиметричного развития челюстей, исправление аномального прикуса, предоперационную подготовку зубов и мимической мускулатуры к операциям. Лечение у ортодонта делится на этапы, соответствующие трем видам прикуса:
Ретенционные мероприятия, завершающие лечение, проводятся для закрепления достигнутого и заканчиваются в 18 лет, когда тело уже практически сформировано. Все это время до достижения совершеннолетия ребенок находится под контролем врачей, в зависимости от степени тяжести заболевания по необходимости ему назначаются лекарства, витамины и разнообразные процедуры. В комплекс лечения обычно входит лечебная гимнастика и работа с сурдопедагогом и психологом.
Альтернативная медицина
Пороки развития черепных и позвоночных структур при синдроме Гольденхара предполагают хирургическое вмешательство, однако, народное лечение может стать дополнительным хорошим подспорьем в реабилитационный период. Хочется только напомнить, что возможность использования народного метода необходимо сначала обговорить с лечащим врачом.
Лечебная гимнастика для исправления прикуса
Эти упражнения нужно делать в течение дня не менее двух раз, каждое из них повторить не менее шести раз:
Полезно также нагружать челюсти, регулярно хорошо пережевывая твердую пищу.
Дермоидные кисты, характерные для синдрома Гольденарха, лечатся только хирургическим путем.
Однако есть и народные методы избавления от кист. Рекомендуется очищать глаза отварами лекарственных трав: например, сделав отвар из цветков василька, листьев подорожника и семян тмина, закапывать по три капли в каждый глаз не менее пяти раз за день.
Можно промывать глаза чайной заваркой или отваром цветков ромашки аптечной: на стакан кипятка – три столовых ложки цветков.
Другой вполне безопасный и витаминный рецепт: смешать 1:1 мед и сок из ягод калины. Первую неделю принимать по одному грамму смеси натощак по утрам (в чайной ложке без верха – восемь граммов меда), на второй неделе – увеличить дозу вдвое, на третьей – еще вдвое, на четвертой – дозировка составляет 10г меда. Затем делают перерыв и повторяют прием в обратной последовательности, начиная с 10г.
При тугоухости тоже применяют лечение травами:
Гомеопатия не сможет заменить оперативные вмешательства, необходимые при множественных пороках развития, присущих синдрому Гольденарха, однако, гомеопатические препараты могут поспособствовать быстрому восстановлению после операции (Арсеникум альбум, Стафизагрия). Отдельные отклонения от нормы, такие как тугоухость (Астэриас рубенс), косоглазие (Танацэтум), новообразования на голове, веках (Крокус сативус, Графитэс, Туя), а также общее состояние больного вполне могут быть сорректированы, особенно после консультации у врача-гомеопата.
[34], [35], [36], [37], [38]
Синдром Гольденхара: что это, причины, симптомы, лечение и операция

Синдром Гольденхара — наследственная односторонняя гипоплазия лица с преимущественным поражением ушной раковины, глаз, носа, зубов, позвоночника. При этом вторая половина лица имеет абсолютно нормальный вид. Это редко встречающееся, генетически обусловленное заболевание отличается неполноценным развитием скелета, мышц, сосудисто-нервных стволов и прочих структур человеческого организма.
Окуло-аурикуло-вертебральная дисплазия — второе название данного недуга. Синдром Гольденхара представляет собой отдельный вид целой группы патологий, для которой характерна задержка физического развития человека. Непосредственной причиной его развития является внутриутробное повреждение жаберных дуг, из которых у плода формируются структуры слухового и жевательного аппарата.
Синдром Гольденхара отличается широким клиническим полиморфизмом. Классическими симптомами заболевания являются: недоразвитие в процессе эмбриогенеза нижней челюсти, резкое нарушение симметрии лица, деформация наружного уха и глаза, нарушение строения позвонков шеи. Возможно полное отсутствие ушной раковины или органа зрения. У больных формируются дермоидные кисты в глазах, развиваются отклонения в работе сердечно-сосудистой, мочеполовой систем и желудочно-кишечного тракта. Клинически это проявляется одышкой, нарушением процесса питания, снижением слуха с пораженной стороны. Синдром Гольденхара не связан с умственной отсталостью. Выявить патологию можно на 20-24 неделе беременности с помощью ультразвуковой диагностики со сканированием в трех измерениях. Большинство случаев синдрома – спорадические.

Впервые патологию описал в 1952 году американский доктор Гольденхар. Синдром получил свое название по фамилии его первооткрывателя. У мальчиков заболевание встречается несколько чаще, чем у девочек. С помощью современных методов диагностики заболевание обнаруживают внутриутробно и решают вопрос о целесообразности сохранения беременности.
Этиология и патогенез
Причины возникновения и механизмы развития синдрома Гольденхара остаются неизвестными. Мнения ученых сходятся на том, что патология относится к наследственным недугам. У пациентов с данным синдромом обнаруживают хромосомные аномалии и мутации генов. Больной ребенок рождается с множественными дефектами лица и патологиями внутренних органов.

Факторы риска рождения больных детей:
Во время замены источника кровоснабжения на участке 1 и 2 жаберных щелей эмбриона происходит кровоизлияние. Разрыв кровеносного сосуда в этом месте приводит к нарушению митотического деления клеток и неправильному формированию основных анатомических структур человеческого организма.
Симптоматика
Первые симптомы патологии можно заметить сразу после рождения ребенка. У больных детей асимметричное лицо, мелкие глазницы, деформированные ушные раковины, недоразвитая нижняя челюсть. Это внешние признаки патологии, которые нередко сопровождаются патологическими изменениями во внутренних органах.

Симптомы со стороны зрительного анализатора:

Клинические признаки при поражении ушей:
Нижняя челюсть у больных недоразвита. Одна половина лица меньше другой, мимические мышцы развиты слабо. Небо имеет вид высокой арки, иногда расщепляется. Ротовая щель при этом слишком широкая, прикус нарушается, появляются добавочные уздечки и расщелины на малом язычке. Нарушается рост зубов. Мягкое небо, губы и гланды деформированы. Имеются трахеопищеводные свищи. Рот большой, один угол выше другого. Недоразвиты скулы, лоб выступает, присутствуют аномалии языка.

У деток с синдромом Гольденхара имеются проблемы с позвоночником: недоразвитие шейного отдела, сколиоз, косолапость, клиновидная форма позвонков, их слияние, наличие полупозвонков, сращение шейных позвонков с затылком, костные аномалии, короткая шея, искривление позвоночника.
Проявлениями со стороны сердечно-сосудистой системы являются симптомы врожденных пороков сердца. Больные дети рождаются с недоразвитием легких, отсутствием органов малого таза, парезом лицевого нерва, гидроцефалией. Они испытывают трудности в обучении. Каждый 10 больной ребенок рождается с поражением ЦНС.
Для синдрома Гольденхара характерно появление симптоматики с одной стороны лица и туловища.
Осложнения и последствия

тяжелое поражение лица и глаз при СГ
Некоторые проявления синдрома Гольденхара не совместимы с жизнью. Ребенок может умереть сразу после рождения.
Осложнения синдрома Гольденхара:
Диагностика
Предварительный диагноз патологии устанавливают сразу после рождения ребенка на основании визуального осмотра. Диагностика включает тщательный опрос родителей и составление анамнеза.
Разнообразные диагностические процедуры позволяют поставить точный диагноз.

Лечение
Лечение синдрома Гольденхара мультидисциплинарное. Дети до трехлетнего возраста наблюдаются у различных специалистов — ЛОР-врача, окулиста, сурдолога, ортопеда и прочих. Детей старше 3 лет направляют к хирургу. В тяжелых случаях для удаления грубых врожденных дефектов сразу после рождения проводят оперативное лечение, а затем комплексную медикаментозную и ортодонтическую терапию.
Хирургическое лечение
Вид оперативного вмешательства определяется тяжестью патологии.

Чтобы предотвратить развитие воспалительных осложнений, во время реабилитации назначают антибиотики и витамины. К остеотропным антибактериальным препаратам относятся «Линкомицин», «Эритромицин», защищенные пенициллины – «Амоксиклав». Для снятия боли назначают анальгетики – «Нурофен», «Ибуклин». Больные дети должны полноценно питаться и принимать поливитаминные комплексы.
Физиотерапия – неотъемлемый компонент лечебного и реабилитационного процесса любых заболеваний. Больным с синдромом Гольденхара проводят УФО, ультразвук, магнитотерапию, лазерное лечение, оксигенотерапию. В терапевтический комплекс входит также лечебная гимнастика, занятия с сурдологом и психологом, слухопротезирование цифровыми имплантируемыми слуховыми аппаратами, контроль слуха и периодические настройки аппаратов. При сколиозе доктора назначают массаж и ношение специальных корсетов.
Лечение у ортодонта проходит в несколько этапов. Первый этап — молочный. Он представляет собой знакомство с патологией. Специалисты объясняют родителям, как правильно ухаживать за полостью рта больного ребенка, предупреждают о возможных осложнениях, проводят аппаратное лечение для исправления дефектов челюсти. Следующий этап — сменный, заключающийся в исправлении прикуса и коррекции имеющихся деформаций во рту. Постоянный этап – замена съемных аппаратов на брекеты и различные фиксаторы. Обычно к 18-летнему возрасту ортодонтическое лечение полностью завершается.
Народная медицина
Наиболее распространенные рецепты народной медицины:
Прогноз

Длительное, раннее и комплексное лечение заболевания в большинстве случаев делает прогноз благоприятным. Пластические операции в некоторых случаях полностью устраняют внешние признаки заболевания. Это позволяет детям посещать общественные места, учиться, а в дальнейшем – работать, полноценно жить, создавать семью и заводить детей. После проведения ряда операций ребенок становится совершенно здоровым.
Если ребенок имеет не только внешние признаки патологии, но и поражение внутренних органов, он получает инвалидность. В случаях с особо тяжелыми проявлениями прогноз неблагоприятен. Длительность жизни больных с синдромом Гольденхара зависит от тяжести поражения жизненно важных органов.
Профилактика
Специфической профилактики синдрома Гольденхара не существует. К общим профилактическим мерам, позволяющим предотвратить это врожденное заболевание, относятся:
Пренатальная диагностика врожденных аномалий заключается в проведении фетоскопии и ультразвукового сканирования эмбриона. Проводят УЗИ на 20-24 неделе беременности. Этот метод является точным на 100%. Он позволяет специалистам понять, стоит ли сохранять беременность.