Мышечные дистонии
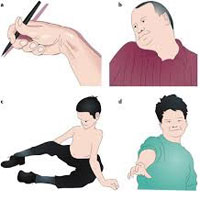
Симптомы дистонии
Мышечная дистония может быть в различных частях тела. Ранние симптомы могут включать в себя ухудшение почерка после написания нескольких строк, судороги в ногах или скованность в мышцах ног, могут возникать неожиданно как «гром среди ясного неба» или возникать после бега или ходьбы на некоторое расстояние. Возможно непроизвольное скручивание шеи, особенно после нагрузки или стресса. Иногда возникает непроизвольное частое моргание глаз, что может приводить к функциональной слепоте. Другие возможные симптомы это тремор и нарушения речи. Начальные симптомы могут быть очень незначительными и могут быть заметны только после длительных нагрузок, стресса или усталости. Со временем симптомы могут стать более явными и стойкими, но, иногда, они могут не прогрессировать.
У некоторых пациентов, симптомы дистонии появляются в детстве, в возрастном промежутке от 5 до 16 лет, чаще всего, в конечностях (ноге или в руке). При генерализованной дистонии дистонические движения могут быстро прогрессировать и вовлекать все конечности и туловище, но скорость прогрессирования обычно заметно замедляется после завершения пубертатного возраста.
У других пациентов, симптомы появляются в конце подросткового или раннего взрослого возраста. В таких случаях, дистония нередко начинается в верхних частях тела, а симптоматика прогрессирует медленно. Дистония, которая начинается в зрелом возрасте, чаще всего, остается фокальной или сегментарной дистонией.
Прогрессирование дистонии проходит несколько стадий. Первоначально дистонические движения преходящие и появляются только во время произвольных движений или стресса. В дальнейшем, у пациентов могут возникать дистонические ненормальные позы и движения во время ходьбы и, в конечном счете, даже в состоянии покоя. Дистонические движения могут со временем привести к стойким физическим дефектам, так как возникают укорочения сухожилий.
При вторичных дистониях вследствие травмы или инсульта у пациентов отмечаются аномальные движения только с одной стороны тела, которые могут появиться сразу после травмы головного мозга (инсульта) или через некоторое время после. Симптомы обычно не прогрессируют и не охватывают другие части тела.
Классификация дистоний
Одна из классификаций дистонии подразделяет их в зависимости от частей тела, вовлеченных в это состояние:
Некоторые типы дистонии выделяются как отдельные синдромы:
Торсионная дистония, которая ранее называлась мышечная деформирующая дистония является редкой формой дистонии, имеет генетическую детерминированность, обычно начинается в детстве и неуклонно прогрессирует. Торсионная дистония приводит к выраженным физическим дефектам и нередко к тяжелой инвалидизации. Исследования генетиков выявили причину этой формы дистонии (у многих пациентов имелись мутации в гене под названием DYT1). Было также отмечено, что этот ген ассоциирован не только с генерализованной дистонией, но и с некоторыми формами фокальной дистонии. Тем не менее, есть данные, что большинство дистоний не связаны с этим дефектом гена и имеют неизвестную причину.
Краниальная дистония это термин, используемый для описания дистонии, которая влияет на мышцы головы, лица и шеи. Оромандибулярная дистония затрагивает мышцы челюсти, губ и языка. Челюсть, может выдвигаться вперед, опускаться или закрываться и возможны нарушения глотания и речи. Спастические дисфония поражает мышцы гортани, которые контролируют речь, что может вызвать нарушения речи, дыхания или хриплость голоса. Синдром Мейга является сочетанием блефароспазма и оромандибулярной дистонии и иногда спастической дисфонии.Спастическая кривошея также иногда классифицируется, как краниальная дистония.
Как правило, ДЗД начинается в детстве или в подростковом возрасте, с прогрессирующим ухудшением процесса ходьбы, а в некоторых случаях и наличием спастичности. При дистонии Segawa, симптомы могут колебаться в течение дня от относительной мобильности утром, с постепенным ухудшением в дневное и вечернее время, а также после физических упражнений. Диагноз ДЗД может быть не выставлен своевременно, так как эта форма дистонии по проявлениям напоминает по симптоматике церебральный паралич. Кроме того, существуют формы дистонии, которые могут иметь четкую генетическую причину: DYT1 дистония является редкой формой доминантно наследуемой генарализованной дистонии, которая может быть вызваны мутацией в гене DYT1. Эта форма дистонии обычно начинается в детстве, сначала влияет на конечности, и неуклонно прогрессирует, часто вызывая инвалидизацию. Поскольку эффекты мутации гена проявляются не всегда, у некоторых людей с наличием мутации этого гена может не быть проявлений дистонии.
В последнее время исследователи выявили еще одну генетическую причину дистонии связанную связано с мутациями в гене DYT6. Дистония вызванная мутацией в гене DYT6 часто проявляется как черепно-лицевая дистония, цервикальная дистонии или дистония руки.
Механизм развития дистонии
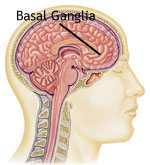 Ученые считают, что дистонии возникают в результате нарушений функционирования в области мозга, которая называется базальные ганглии, где происходит обработка информации от импульсов, которые поступают в мозг при сокращениях мышц. Ученые предполагают, что происходит нарушение выработки организмом определенной группы химических веществ (нейротрансмиттеров), которые позволяют клеткам мозга взаимодействовать друг с другом. Некоторые из этих нейротрансмиттеров включают:
Ученые считают, что дистонии возникают в результате нарушений функционирования в области мозга, которая называется базальные ганглии, где происходит обработка информации от импульсов, которые поступают в мозг при сокращениях мышц. Ученые предполагают, что происходит нарушение выработки организмом определенной группы химических веществ (нейротрансмиттеров), которые позволяют клеткам мозга взаимодействовать друг с другом. Некоторые из этих нейротрансмиттеров включают:
Приобретенные дистонии, которые также называются вторичные дистонии, являются результатом влияния экзогенных факторов или заболеваний, приводящих к повреждению базальных ганглиев. Родовая травма (в частности, из-за гипоксии мозга), некоторые инфекции, реакция на некоторые лекарства, тяжелые металлы или отравления окисью углерода, травмы, или инсульт могут привести к появлению дистонических симптомов. Дистонии также могут быть симптомами других заболеваний, некоторые из которых могут быть наследственными.
Около 50% случаев дистонии не имеют никакой связи с заболеваниями или травмами и называются первичной или идиопатической дистонией.
Некоторые случаи первичной дистонии могут иметь различные типы наследственных паттернов.
Лечение
 В настоящее время не существует медикаментов, которые могут предотвратить развитие дистонии или замедлить прогрессирование. Однако существует несколько вариантов лечения, которые могут облегчить некоторые симптомы дистонии, поэтому врачи могут подобрать каждому пациенту определенную тактику лечения, основанную на конкретных симптомах.
В настоящее время не существует медикаментов, которые могут предотвратить развитие дистонии или замедлить прогрессирование. Однако существует несколько вариантов лечения, которые могут облегчить некоторые симптомы дистонии, поэтому врачи могут подобрать каждому пациенту определенную тактику лечения, основанную на конкретных симптомах.
Хирургические методы лечения в некоторых случаях могут быть рекомендованы отдельным пациентам, когда лекарства оказываются не эффективны или побочные эффекты являются слишком серьезными. В отдельных случаях при выраженной генерализованной дистонии проводится хирургическая деструкция части таламуса, глубокой структуры мозга контролирующей движения. Нарушение речи является основным риском такой процедуры, так как таламус располагается около структур головного мозга, контролирующих речь. Хирургическая денервация мышц иногда помогает при фокальной дистонии, в том числе при блефароспазме, спазматической дисфонии и кривошее. Но результаты таких денерваций не очень обнадеживающие в долгосрочной перспективе.
Использование материалов допускается при указании активной гиперссылки на постоянную страницу статьи.
Мышечная дистония
Мышечная дистония – это синдром, при котором у пациента наблюдается неконтролируемое сокращение мышц. В результате больной вынужден принимать неестественную позу, если затронуты конечности, то они необычно изогнуты. Такой диагноз, как мышечная дистония, очень часто ставят детям в раннем возрасте. Это связано с тем, что патология в подавляющем большинстве случаев проявляется сразу же после рождения. Реже от мышечной дистонии страдают молодые, взрослые и пожилые пациенты.
Причины и предпосылки болезни
Главная причина – это родовая травма или врожденные травмы головного мозга. Если во время беременности или родов в головном мозге нарушается кровообращение, возникает кислородное голодание.
У взрослых установить причину болезни сложнее. Иногда мышечная дистония развивается на фоне приема различных лекарственных средств, после черепно-мозговых травм, может поздно проявиться дистония, вызванная дегенеративными наследственными заболеваниями. Очень часто взрослым пациентам ставят диагноз идиопатическая дистония – то есть неуточненной этиологии.
Разновидности синдрома
Как было отмечено ранее, мышечная дистония – это отклонения в функционировании отдельных мышц или мышечных групп, которые проявляются чрезмерной скованностью, ограничением двигательной активности, неравномерным тонусом. В связи с этим выделяют две основные формы заболевания: гипертонус и гипотонус. При гипертонусе отмечено повышенное напряжение мышц, при гипотонусе – пониженное.
Заболевание классифицируют на несколько форм по локализации и степени распространения:
Фокальные дистонии распространены сильнее, чем остальные формы. Их, в свою очередь, также несколько типов:
Фокальные дистонии чаще всего встречаются у взрослых людей трудоспособного возраста. Симптомы заболевания снижают качество жизни, способствуют снижению работоспособности, повышают риск социальной дезадаптации и инвалидизации. Нередко синдром приводит к образованию функциональных дефектов – например, расстройства зрения при блефароспазме вплоть до функциональной слепоты, сложности с удержанием головы при цервикальной форме заболевания. Поэтому лечение направлено не только на устранение симптоматики, но и на восстановление и поддержание функций организма.
Симптомы и проявления
У детей симптомы отличаются от взрослых. Так, в раннем возрасте родители могут заметить односторонние или полностью ассиметричные симптомы. Например, ребенок переворачивается только на одну сторону и полностью игнорирует другую. Или поворачивает голову только на одну сторону.
У взрослых основной симптом – неестественная походка, аномальные, нетипичные позы тела и положения конечностей. Расстройство проявляется в основном симптомами со стороны скелетной мускулатуры. Иногда позы немного необычные, а иногда кажутся неестественными даже на первый взгляд.
У взрослых дистония может привести к инвалидности и полной нетрудоспособности, особенно если в роли причины выступает нарушение мозгового кровообращения.
Поэтому важно своевременно обращаться к врачу. Единственный способ сохранить трудоспособность – вовремя начать лечение.
Мышечная дистония

Мышечная дистония — это синдром поражения ЦНС, проявляющийся несогласованными аритмичными изменениями тонуса различных групп мышц. Клинически характеризуется фокальными или генерализованными непроизвольными фиксированными позами или двигательными актами. Диагностика проводится по клиническим данным о наличии дистонических феноменов. Инструментальные обследования (МРТ, УЗДГ, ЭНМГ) направлены на поиск причинного заболевания. Консервативная терапия включает кинезиотерапию, назначение фармпрепаратов, локальное введение ботулотоксина, физиотерапевтические воздействия. При генерализованных формах возможно нейрохирургическое лечение.
МКБ-10

Общие сведения
Мышечное напряжение (тонус) необходимо для поддержания позы тела, осуществления движений. В отличие от сниженного (гипотонус) и повышенного (гипертонус) напряжения мышц, мышечная дистония (МД) обозначает нарушение адекватного соотношения тонуса отдельных мышц или мышечных групп. МД не является нозологической единицей, представляет собой синдром, встречающийся при различных поражениях центральной нервной системы. По данным европейских исследований, мышечная дистония встречается в странах Западной Европы с частотой 11,2 случая на 100 тыс. населения. Начало проявлений возможно в любом возрасте. Более ранний дебют симптоматики ведёт к более быстрому прогрессированию синдрома с последующей генерализацией процесса.
Причины
Этиофакторами МД могут выступать различные поражения головного мозга, распространяющиеся на структуры ответственной за регуляцию тонуса экстрапирамидной системы. У детей первого года жизни мышечная дистония развивается преимущественно вследствие нарушений развития нервной системы во внутриутробном периоде или её повреждений в процессе родов. К возможным причинам относят гипоксию плода, внутриутробные инфекции, токсические воздействия (курение, приём алкоголя, наркомания, медикаментозное лечение беременной), родовую травму новорожденного. У детей старшего возраста и взрослых выделяют следующие основные этиологические факторы:
Патогенез
Указанные выше этиофакторы вызывают дисфункцию многоуровневой системы регуляции мышечного напряжения. В результате возникает спонтанная импульсация, чрезмерно активирующая определённые мышцы. Активированная мышечная группа входит в состояние тонического сокращения, что приводит к насильственному движению с последующим застыванием. В зависимости от локализации подверженной патологической импульсации мышечной группы возможен непроизвольный поворот головы, скручивание туловища, зажмуривание глаза, дистонический тризм и т. п. Чередование избыточного напряжения мышц-антагонистов лежит в основе гиперкинезов — непроизвольных двигательных актов различной амплитуды и скорости. Патогенетический механизм развития многих МД продолжает изучаться.
Классификация
По локализации дистонических феноменов выделяют краниальную, параорбитальную, оромандибулярную, цервикальную, фарингеальную, торсионную и прочие формы МД. По этиологии дистонический синдром классифицируют на первичный (наследственный, идиопатический), вторичный (приобретённый). В основу следующей классификации легла распространённость патологического процесса на мышечные группы. В соответствии с данным критерием выделяют следующие формы МД:
По мере развития дистонического синдрома усугубляется выраженность патологических изменений. В связи с этим выделяют 4 степени выраженности МД. В клинической практике они оцениваются как стадии развития дистонии.
Симптомы мышечной дистонии
Основные клинические проявления: дистонические позы и двигательные акты. Полиморфизм симптоматики обусловлен различной локализацией процесса, силой, скоростью и частотой мышечных сокращений. Общей закономерностью для дистонических феноменов является их стереотипный характер, возникновение в процессе выполнения произвольных двигательных актов, усиление при переутомлении, депривации сна, стрессе, уменьшение после сна, отдыха, в гипнотическом состоянии.
В большинстве случаев мышечная дистония манифестирует фокальными формами. Писчий спазм проявляется спастическим сокращением мышц кисти, что делает невозможным дальнейшее написание текста. Идиопатический блефароспазм характеризуется сокращением круговой мышцы глаза, приводящим к зажмуриванию. Оромандибулярная форма имеет несколько вариантов: тризм, насильственное открывание рта, вытягивание губ, высовывание языка. Цервикальная МД характеризуется насильственным поворотом, наклоном головы, фарингеальная — нарушением глотания, торсионная — скручивающими туловище поворотами. Приспосабливаясь к дистоническим приступам, пациенты вырабатывают привычные корригирующие жесты и движения, уменьшающие выраженность мышечных нарушений.
У грудничков мышечная дистония проявляется неестественными позами, поворотом головы всегда в одну сторону, переворачиванием только через один бок, отставанием статико-моторного развития. Во многих случаях фокальные проявления постепенно трансформируются в сегментарные, распространяются на другие участки тела, спустя несколько лет переходят в генерализованную МД. Чем раньше произошёл дебют дистонии, тем быстрее возникает генерализация. Длительное изолированное течение фокальных дистоний наблюдается у больных с манифестацией клинической симптоматики после 25-35 лет.
Осложнения
Прогрессирующая мышечная дистония значительно нарушает двигательную сферу больного, затрудняет профессиональную и бытовую деятельность. Со временем выполнение профессиональных обязанностей становиться невозможным, пациент переходит на инвалидность. Цервикальная форма осложняется развитием кривошеи. При торсионной МД развиваются искривления позвоночника, на поздних стадиях возможны дыхательные расстройства. Блефароспазм приводит к возникновению энтропиона, сухости переднего сегмента глаза. В ряде случаев отмечается возникновение невритов и компрессионных невропатий периферических нервов конечности, осуществляющей корригирующие дистонию движения.
Диагностика
Диагноз устанавливается врачом-неврологом на основании клинических данных с учетом жалоб на дистонические проявления и результатов осмотра. Дальнейшие диагностические обследования направлены на определение причины возникновения дистонического синдрома, верификацию базового заболевания. Основными этапами диагностики являются:
Дифференциальная диагностика проводится с боковым амиотрофическим склерозом, нейроакантоцитозом, болезнью Мачадо-Джозефа. Отличительным признаком МД является наличие корригирующего жеста, который отсутствует при иных экстрапирамидных расстройствах. Отсутствие мышечных атрофий позволяет отличить дистонический синдром от болезни двигательного нейрона. Для нейроакантоцитоза характерно наличие невротических расстройств, выявление в анализе крови изменённых эритроцитов (акантоцитов).
Лечение мышечной дистонии
Терапия направлена на уменьшение симптоматики, улучшение самочувствия пациента. Её составляющей является исключение усугубляющих дистонию факторов (переутомления, избыточного мышечного напряжения, недостаточного сна, стрессовых ситуаций). С целью достижения лучшего результата в лечении используются сочетание нескольких из указанных ниже методик:
Хирургическое лечение показано при выраженной генерализованной МД на фоне недостаточной эффективности консервативных методов. Наиболее часто применяется стереотаксическая деструкция вентролатерального таламического ядра. Методики хирургического лечения различных форм заболевания продолжают разрабатываться.
Прогноз и профилактика
Течение вторичного дистонического синдрома зависит от характера основного заболевания. Прогноз идиопатических форм тем серьёзнее, чем раньше произошла манифестация клинических проявлений. Своевременно начатая комплексная терапия позволяет намного улучшить качество жизни пациента, отсрочить инвалидизацию. Профилактика состоит в исключении неблагоприятных воздействий на плод в период внутриутробного развития, адекватном выборе способа родоразрешения, предупреждении травматизма, инфекционных и онкологических поражений ЦНС, тщательном планировании сроков и дозировок при проведении медикаментозной терапии.
Мышечные дистонии причины, симптомы, методы лечения и профилактики
Мышечные дистонии — двигательные расстройства, вызванные поражением центральной нервной системы, для которых характерны непроизвольные сокращения отдельных мышц или мышечных групп. Проявляются патологическими позами, двигательными актами, возникающими помимо воли больного. Начало возможно в любом возрасте. Лечение и диагностику мышечных дистоний проводят специалисты в области неврологии.

Причины мышечной дистонии
Причины нарушения тонуса мышц разнообразны, чаще связаны с поражением головного мозга. У детей первого года жизни неврологические расстройства возникают вследствие недоразвития нервной системы во внутриутробном периоде или повреждении ее во время родов. Спровоцировать дистонию у новорожденного способно кислородное голодание плода, родовая травма, алкогольная, никотиновая, лекарственная интоксикация беременной.
У детей старшего возраста и взрослых выделяют другие причины мышечной дистонии, а именно:
Статью проверил

Дата публикации: 24 Марта 2021 года
Дата проверки: 24 Марта 2021 года
Дата обновления: 08 Декабря 2021 года
Содержание статьи
Симптомы мышечной дистонии
В ответ на расстройство механизмов регуляции мышечного тонуса возникает спонтанная импульсация, чрезмерно активирующая определенную группу мышц. Избыточный характер патологической двигательной активности выражается в насильственных движениях — гиперкинезах. В большинстве случаев они сочетаются с другими симптомами, типичными для первичного заболевания. Это:
Стадии развития мышечной дистонии
В соответствии с выраженностью двигательных расстройств выделяют четыре стадии развития дистонии мышц:
Разновидности
По происхождению выделяют первичный (наследственный) и вторичный (приобретенный) тип дистонии. В соответствии с вовлечением в патологический процесс мышечных групп существует несколько форм мышечной дистонии:
Диагностика
Диагноз основан на проявлениях мышечной дистонии с учетом жалоб пациента, данных неврологического осмотра. В диагностике некоторых заболеваний необходима консультация кардиолога, онколога, травматолога, генетика. Для подтверждения или опровержения вторичных процессов развития дистонического синдрома в план диагностики включают аппаратные методы исследования, как:
В сети клиник ЦМРТ диагностику причин нарушения тонуса мышц проводят следующими способами:
Диагностика и лечение экстрапирамидных гиперкинезов
Экстрапирамидные гиперкинезы относятся к числу расстройств, которые не столько угрожают жизни, сколько «разрушают» ее, значительно ограничивая функциональные возможности пациентов, приводя их к психологической и социальной изоляции. Длительное время ре
Экстрапирамидные гиперкинезы относятся к числу расстройств, которые не столько угрожают жизни, сколько «разрушают» ее, значительно ограничивая функциональные возможности пациентов, приводя их к психологической и социальной изоляции. Длительное время результаты лечения экстрапирамидных гиперкинезов вызывали лишь разочарование как у самих пациентов, так и у врачей. Но в последние десятилетия ситуация начала меняться. Появились более четкие критерии диагностики различных вариантов экстрапирамидных гиперкинезов, существенно расширились возможности лечения, как за счет появления новых методов, так и за счет более рационального применения ранее существовавших. И если мы до сих пор в подавляющем большинстве случаев не можем кардинально излечить гиперкинез, то, по крайней мере, способны существенно улучшить качество жизни многих пациентов. В данной статье рассмотрены современные подходы к диагностике и лечению наиболее частых вариантов экстрапирамидных гиперкинезов.
Определение и классификация экстрапирамидных гиперкинезов
Экстрапирамидные гиперкинезы (или дискинезии) — это непроизвольные (насильственные) избыточные движения, обусловленные поражением базальных ганглиев и связанных с ними структур, условно объединяемых в экстрапирамидную систему [9]. Экстрапирамидные гиперкинезы следует отличать от более редких периферических гиперкинезов, связанных с поражением или дисфункцией периферических нервов (например, лицевого гемиспазма, синдрома «болезненных ног (рук) — движущихся пальцев», тетания и др.), а также от психогенных гиперкинезов, являющихся соматическим выражением того или иного психического заболевания.
К основным экстрапирамидным гиперкинезам относят тремор, дистонию, хорею, атетоз, баллизм, тики, миоклонию, акатизию [11]. Традиционно считается, что каждый гиперкинез имеет свой неповторимый двигательный рисунок, в основе которого лежит уникальный патофизиологический механизм. Отчасти это действительно так. Тем не менее накопленный нами опыт позволяет говорить не столько об отдельных, дискретных синдромах, сколько о едином спектре (континууме) синдромов, в котором наряду с изолированными формами широко представлены переходные или комбинированные формы, что существенно затрудняет их синдромальную диагностику и выбор правильного лечения.
Справедливо мнение, что гиперкинезы «сопротивляются» жесткой вербальной категоризации, и их значительно проще узнать, чем описать. Ситуация усложняется еще и тем обстоятельством, что один и тот же гиперкинез в разных частях тела может выглядеть по-разному. В связи с этим распознавание гиперкинезов, особенно в сложных или переходных случаях, невозможно без выделения ограниченного числа ключевых признаков. По нашему мнению, особенно важное значение имеют три признака: двигательный рисунок, временной рисунок, характер возникновения.
По двигательному рисунку гиперкинезы могут быть разделены на три основные группы:
По временному рисунку гиперкинезы могут быть разделены на две группы:
По характеру возникновения непроизвольные гиперкинезы могут быть разделены на четыре основные группы:
Феноменологические особенности основных форм экстрапирамидных гиперкинезов, в сравнении с психогенными гиперкинезами, представлены в таблице.
Общие принципы диагностики экстрапирамидных гиперкинезов
Распознавание того или иного экстрапирамидного синдрома — только отправная точка сложной диагностической работы, итогом которой может быть установление нозологического диагноза.
Диагностика экстрапирамидного синдрома включает три последовательных этапа.
С нозологической точки зрения в рамках любого экстрапирамидного гиперкинеза могут быть выделены три основные формы.
Большинство случаев экстрапирамидных гиперкинезов имеют первичный (идиопатический) характер, однако их диагностика требует исключения других, прежде всего вторичных, форм гиперкинезов, особенно связанных с курабельными заболеваниями (такими, как опухоли или эндокринопатии), а также курабельных форм мультисистемных дегенераций, в первую очередь гепатолентикулярной дегенерации (болезни Вильсона–Коновалова). Подобные случаи в клинической практике встречаются редко, но именно они должны быть исключены в первую очередь. Исключение вторичной природы гиперкинеза может потребовать дополнительного инструментального (КТ или МРТ головного мозга, ЭЭГ) либо лабораторного исследования. Следует помнить, что любой экстрапирамидный синдром, впервые проявившийся в возрасте до 50 лет, служит основанием для исключения гепатолентикулярной дегенерации (для этого требуется как минимум анализ крови на церулоплазмин и исследование роговицы с помощью щелевой лампы с целью обнаружения пигментного кольца Кайзера–Флейшера) [12].
Наконец, в каждом случае гиперкинеза следует подумать и о том, что он может иметь психогенную природу. В прошлом большинство случаев гиперкинезов нередко рассматривались как психогенные расстройства. Этому способствовали вариабельность и динамичность проявлений экстрапирамидных гиперкинезов, их зависимость от движений, позы, эмоционального состояния пациента, нередкое присутствие у пациентов с первичными формами гиперкинезов аффективных расстройств. В настоящее время очевидно, что психогенные гиперкинезы встречаются редко, но тем более важным представляется их своевременное выявление, позволяющее проводить целенаправленное лечение и как минимум избавляющее пациента от ненужной,
а иногда и опасной для него терапии.
В пользу психогенной природы гиперкинеза могут свидетельствовать: острое начало, последующее волнообразное течение с периодами длительных спонтанных ремиссий, непостоянство гиперкинеза, причудливость его рисунка, обычно не соответствующего характерным формам экстрапирамидных гиперкинезов, ослабление при отвлечении внимания, стойкая реакция на плацебо, полная резистентность к стандартной терапии, наличие других псевдоневрологических симптомов с феноменом селективной несостоятельности, выраженных аффективных расстройств, сопровождающихся множественными соматоформными жалобами, наличие рентной ситуации (в которой больной извлекает моральную или, реже, материальную выгоду из своего заболевания) и т. д. [1].
Ниже более подробно рассмотрены подходы к диагностике и лечению четырех наиболее часто встречающихся форм экстрапирамидных гиперкинезов: тремора, дистонии, хореи и тиков.
Тремор
Тремор (дрожание) — самый частый экстрапирамидный гиперкинез, характеризующийся непроизвольными ритмичными колебательными движениями части тела (чаще всего конечностей и головы) или всего тела, которые упорядочены во времени и пространстве. Феноменологически выделяют два основных типа тремора: тремор покоя и тремор действия (акционный тремор). Тремор покоя характерен для синдрома паркинсонизма, и прежде всего болезни Паркинсона.
Тремор действия подразделяют на постуральный, возникающий при удержании определенной позы (например, вытянутых рук), кинетический, появляющийся при движении (в том числе при приближении к цели — так называемый интенционный тремор), изометрический — при изометрическом мышечном сокращении (например, при сжимании кисти в кулак). К особым формам тремора относятся ортостатический тремор, развивающийся при переходе в вертикальное положение и стоянии, а также селективный кинетический тремор (возникающий только при определенных движениях, например при письме — писчий тремор).
Основной формой первичного тремора является эссенциальный тремор (ЭТ), представляющий собой самостоятельное заболевание, преимущественно проявляющееся постурально-кинетическим тремором рук, реже головы, голосовых связок, ног, туловища. Более чем в половине случаев заболевание носит семейный характер. Анализ семейных случаев указывает на аутосомно-доминантный тип наследования, однако установить генетический дефект удалось лишь в отдельных случаях [4]. Не исключено, что спорадические случаи, как правило, проявляющиеся в более позднем возрасте (часто после 60 лет), носят мультифакторный характер и связаны как с генетическим дефектом, так и с воздействием неидентифицированных внешних факторов. ЭТ начинается постепенно, обычно с постурального дрожания в руках, которое может быть как симметричным, так и асимметричным. Со временем амплитуда и распространенность тремора нарастают, тогда как его частота снижается (от 6–8 до 4 Гц). Резко выраженный постуральный тремор может сохраняться и в покое. Помимо косметического дефекта, тремор может нарушать функцию верхних конечностей: больным становится все труднее принимать пищу, писать, играть на музыкальных инструментах, выполнять другие тонкие действия. Однако в некоторых случаях, несмотря на существование заболевания в течение нескольких десятилетий, инвалидизации не наступает.
Другие неврологические проявления обычно отсутствуют, но примерно у трети больных выявляются минимальные проявления мозжечковой атаксии (например, нарушения тандемной ходьбы), минимальная гипомимия, иногда миоклония и фокальная дистония. У больных ЭТ чаще, чем в среднем в популяции, наблюдаются артериальная гипертензия, нейросенсорная тугоухость, когнитивные нарушения.
Как особые варианты ЭТ рассматривают первичный ортостатический тремор, изолированный тремор головы, а также тремор, возникающий при письме (писчий тремор). Последний занимает промежуточное положение между тремором и дистонией. Изолированный тремор головы, возникающий на фоне ее дистонической позы, как правило, представляет собой дистонический тремор, являясь вариантом фокальной дистонии (см. ниже).
ЭТ необходимо также дифференцировать с усиленным физиологическим тремором, возникающим при волнении, утомлении, под действием холода и некоторых лекарственных средств, при абстинентном синдроме, тиреотоксикозе, гипогликемии, интоксикациях; мозжечковым (преимущественно интенционным) тремором, тремором Холмса (асимметричным крупноразмашистым дрожанием, которое представляет собой комбинацию постурального и кинетического тремора с тремором покоя и возникает при очаговых поражениях среднего мозга или таламуса), тремором при полиневропатиях [3].
До сих пор в клинической практике возникают большие сложности при дифференциальной диагностике ЭТ с болезнью Паркинсона. Для последней, в отличие от ЭТ, характерны присутствие других симптомов паркинсонизма, прежде всего выраженной гипокинезии, более быстрое прогрессирование, выраженная асимметрия проявлений, преобладание тремора покоя, отсутствие тремора головы, иная последовательность вовлечения конечностей (рука–ипсилатеральная нога–контралатеральные конечности; при ЭТ: рука–контралатеральная рука–ноги), лечебный эффект противопаркинсонических средств.
К сожалению, в настоящее время возможности предупредить или хотя бы замедлить прогрессирование заболевания не существует. Тем не менее значительная часть пациентов с ЭТ не нуждается ни в каком ином лечении, кроме как в рациональной психотерапии, заключающейся в разъяснении доброкачественной природы заболевания. Если тремор существенно нарушает функцию рук, его можно частично уменьшить почти у 2/3 больных с помощью средств первого ряда — β-блокаторов (пропранолол, 60–360 мг/сут) и примидона (гексамидин, 125–500 мг/сут). Выбор препарата производят исходя из риска побочного действия, сопутствующих заболеваний и индивидуальных особенностей пациентов. У молодых больных, а также пациентов с артериальной гипертензией чаще применяют β-блокаторы, тогда как у пожилых пациентов, особенно чувствительных к побочному действию пропранолола на сердечно-сосудистую систему, более целесообразен прием примидона, который к тому же в большинстве случаев достаточно применять всего 1 раз в сутки — перед сном [5]. Чтобы улучшить переносимость примидона, его терапевтическая доза подбирается путем медленного титрования. После достижения эффективной дозы побочные эффекты наблюдаются редко. В резистентных случаях возможна комбинация двух препаратов первого ряда либо их назначение в сочетании с препаратами второго ряда, к которым относятся клоназепам и алпразолам (особенно эффективны при кинетическом треморе и треморе головы), фенобарбитал, антагонисты кальция (флунаризин, нимодипин), габапентин, топирамат и теофиллин. При треморе головы и голосовых связок единственный метод, дающий гарантированный эффект, — регулярные инъекции ботулотоксина. В наиболее резистентных случаях прибегают к клозапину или проводят стереотаксическое нейрохирургическое вмешательство на таламусе.
Коррекция усиленного физиологического тремора включает прекращение действия провоцирующего фактора, применение β-блокаторов (например, пропранолола). При мозжечковом треморе, обычно плохо поддающемся лечению, обычно назначают ГАМКергические препараты (клоназепам, вальпроевую кислоту, баклофен, габапентин), карбамазепин, пропранолол, примидон, амантадин, практикуется также утяжеление конечности с помощью браслета. В наиболее тяжелых случаях возможно применение изониазида. При треморе Холмса иногда эффективны холинолитики, препараты леводопы, агонисты дофаминовых рецепторов, клоназепам, клозапин, комбинация вальпроевой кислоты и пропранолола, введение ботулотоксина.
Дистония
Дистония — синдром, характеризующийся медленными (тоническими) или повторяющимися быстрыми (клонико-тоническими) движениями, вызывающими вращение (отсюда термин «торсионная дистония» — от лат. torsio — вращение, скручивание), сгибание или разгибание туловища и конечностей с формированием патологических поз.
В отличие от более быстрого и хаотичного хореического гиперкинеза (см. ниже) рисунок дистонического гиперкинеза более стереотипен и упорядочен [1, 2]. Дистонические феномены многообразны и включают преходящие дистонические спазмы, которые иногда бывают столь быстрыми, что напоминают миоклонию (при «клонической» форме дистонии) или относительно ритмичный дистонический тремор, обычно усиливающийся при попытке больного преодолеть дистоническую позу.
Характерная особенность дистонического гиперкинеза — возникновение или усиление при произвольных движениях. Дистоническая поза первоначально имеет преходящий характер, возникает лишь при определенном движении, но постепенно становится постоянной, сохраняясь и в покое. Эта эволюция дистонии весьма характерна, наряду с другими проявлениями динамичности гиперкинезов: улучшение после сна, влияние корригирующих жестов и изменений позы (дистония часто усиливается в вертикальном положении и уменьшается в горизонтальном), наличие парадоксальной кинезии (уменьшение гиперкинеза при изменении привычного двигательного стереотипа), колебания симптоматики, влияние эмоционального состояния. Признаки динамичности гиперкинезов, включая возможность кратковременной произвольной коррекции патологической позы, позволяют отличить дистонию от заболеваний скелетно-мышечной системы, вызывающих более фиксированные изменения позы (псевдодистонии).
По распространенности гиперкинеза выделяют:
Почти 90% случаев составляет первичная (идиопатическая) дистония, которая проявляется только дистоническим гиперкинезом и имеет наследственный характер, но бывает представлена как семейными, так и спорадическими случаями. При раннем дебюте (до 15 лет) дистония обычно имеет четко наследственный характер, часто начинается с одной ноги, а затем генерализуется, вовлекая туловище. При более позднем начале (после 21 года) дистония чаще оказывается представлена спорадическими случаями, первично вовлекает мышцы верхней части тела, а в дальнейшем чаще остается фокальной. В классическом варианте она наследуется по аутосомно-доминантному типу и связана с мутацией в локусе DYT1 на 9-й хромосоме, кодирующем белок торсин А. Реже встречаются другие варианты генерализованной дистонии с наследованием по аутосомно-рецессивному или рецессивному, сцепленному с Х-хромосомой типу [4].
Фокальные формы встречаются примерно в 10 раз чаще, чем генерализованные. К числу частых фокальных вариантов относятся краниальная дистония, включающая блефароспазм и оромандибулярную (орофациальную) дистонию, и цервикальная дистония. Сочетание орофациальной дистонии с гиперкинезом других мышц лица, в том числе с блефароспазмом и дистонией мышц шеи (сегментарная краниоцервикальная дистония), обозначают как синдром Мейжа.
В некоторых семейных случаях сегментарной краниоцервикальной дистонии, при которой имеется сочетание спастической кривошеи с блефароспазмом и орофациальной дистонией, выявляется генетический дефект (ген DYT6 на 8-й хромосоме). В семейных случаях чисто цервикальной дистонии выявлен патологический ген DYT7 на 18-й хромосоме. Однако причина большинства случаев цервикальной дистонии остается неясной (идиопатическая цервикальная дистония) [4].
В последние годы выделена группа заболеваний, условно обозначаемых как дистония-плюс, при которых дистонический гиперкинез сопровождается другими экстрапирамидными расстройствами, в частности симптомами паркинсонизма (дистония/дистония-паркинсонизм, чувствительная к L-ДОФА, или ДОФА-зависимая, дистония, или болезнь Сегавы) или миоклонией (миоклоническая дистония).
Вторичная (симптоматическая) дистония составляет не более 5–10% случаев дистонии. Чаще всего она возникает после очагового повреждения базальных ганглиев или таламуса (например, при инсульте), развиваясь спустя несколько месяцев, иногда на фоне регресса гемипареза («отставленная» дистония). Дистония конечности изредка возникает на фоне тяжелой рефлекторной симпатической дистрофии, развившейся после периферической травмы. Важнейшей причиной вторичной дистонии служит воздействие лекарственных средств, прежде всего нейролептиков, метоклопрамида, препаратов леводопы.
Наиболее курабельны ДОФА-зависимые формы дистонии (например, болезнь Сегавы), при которых эффективны малые дозы препаратов леводопы (суточную дозу — от 0,25 до 1,5 табл. накома или мадопара 250 — назначают в один или два приема). Поскольку ДОФА-зависимую дистонию не всегда удается дифференцировать клинически, препараты леводопы целесообразно испробовать во всех случаях генерализованной дистонии, развившейся в детском и юношеском возрасте.
В целом при генерализованной дистонии можно рекомендовать назначение препаратов в следующей последовательности: препараты леводопы (в детском и юношеском возрасте); холинолитики (обычно в высокой дозе, например до 100 мг циклодола в сутки); баклофен; клоназепам и другие бензодиазепины; карбамазепин (финлепсин); препараты, истощающие запасы дофамина в пресинаптических депо (резерпин); нейролептики — блокаторы дофаминовых рецепторов (галоперидол, пимозид, сульпирид, фторфеназин); комбинация из перечисленных средств (например, холинолитика с резерпином и нейролептиком).
Следует отметить, что во многих случаях эффекта удается добиться лишь при применении высоких доз лекарственных средств. В резистентных случаях прибегают к стереотаксическим операциям на бледном шаре или таламусе.
Наиболее эффективный метод лечения фокальных дистоний — инъекции ботулотоксина (ботокса или диспорта) в мышцы, вовлеченные в гиперкинез. Ботулотоксин вызывает частичный парез этих мышц и тем самым устраняет дистонию на 3–6 мес, после чего инъекцию приходится повторять [7]. Возможности лекарственных средств весьма ограничены. При цервикальной дистонии эффекта иногда удается добиться с помощью клоназепама, баклофена или нейролептиков. При блефароспазме более эффективны клоназепам и холинолитики, при оромандибулярной дистонии — баклофен и холинолитики, при «писчем спазме» — холинолитики. В части случаев некоторого улучшения, которое носит скорее субъективный характер, можно добиться путем воздействия на мышцы, участвующие в гиперкинезе, с помощью различных физиотерапевтических процедур, а также применяя метод биологической обратной связи или специальную гимнастику. В резистентных случаях прибегают к периферической денервации мышц.
Хорея
Хорея характеризуется непрерывным потоком быстрых хаотичных, нерегулярных во времени и по амплитуде мультифокальных движений. Гиперкинез чаще всего вовлекает дистальные отделы конечностей, мимические мышцы, иногда мышцы глотки, гортани, туловища. Насильственные движения напоминают гримасничанье, кривляние, нарочитые ужимки, танцевальные движения (греч. choreia — пляска) [8].
К наиболее частым формам хореи относится болезнь Гентингтона (БГ) — наследственное заболевание, передающееся по аутосомно-доминантному типу, связанное с прогрессирующей дегенерацией нейронов подкорковых ядер и коры и проявляющееся главным образом сочетанием хореи с деменцией [4]. Тем не менее хорея — не единственное, а в ряде случаев и не основное проявление заболевания, поэтому термин «болезнь Гентингтона» предпочтительнее, чем термин «хорея Гентингтона». Генетический дефект при БГ выявлен на 4-й хромосоме и заключается в увеличении количества повторов («экспансии») одного из тринуклеотидных фрагментов в зоне ДНК, кодирующей белок гентингтин. В конечном итоге это предопределяет особую уязвимость и преждевременную гибель определенных популяций нейронов полосатого тела, прежде всего хвостатого ядра.
БГ обычно проявляется на 4–5-м десятилетии жизни и в дальнейшем неуклонно прогрессирует. Хорея обычно начинается с дистальных отделов конечностей, затем постепенно генерализуется и нарушает произвольные движения. Больные не могут долго держать высунутым язык или сжимать кисть в кулак, походка становится неустойчивой, «танцующей», иногда замедленной, напряженной. Со временем непроизвольные движения все более приобретают дистонический характер, присоединяются гипокинезия и ригидность, оживление рефлексов, грубая постуральная неустойчивость, приводящая к частым падениям. Уже на ранней стадии часто наблюдается выраженная дизартрия с замедленной аритмичной речью; дисфагия появляется на более поздней стадии и бывает причиной аспирации, ведущей к асфиксии или пневмонии. Психические расстройства многообразны и бывают представлены нарастающим когнитивным дефицитом, депрессией с нередкими суицидальными попытками, навязчивыми и фобическими расстройствами, психотическими нарушениями [9].
Малая хорея, являющаяся осложнением ревматизма и в прошлом составляющая значительную часть случаев вторичной хореи, в последние годы возникает исключительно редко. В связи с этим при возникновении хореи в детском или юношеском возрасте важно исключать иные причины синдрома: сосудистую хорею, системную красную волчанку, антифосфолипидный синдром и др. У пожилых людей хорея чаще бывает вызвана полицитемией, заболеваниями печени, последствиями инсульта.
При умеренном гиперкинезе с нейролептиками могут конкурировать средства, блокирующие глутаматергические рецепторы (например, амантадин или мемантин), некоторые антиконвульсанты (например, топирамат), а также симпатолитики (например, резерпин), истощающие запасы катехоламинов (в том числе дофамина) в депо пресинаптических терминалей. В некоторых случаях возможна комбинация лечебных средств, в частности нейролептика с антиглутаматергическими средствами, антиконвульсантами и симпатолитиками. Согласно некоторым данным длительный прием коэнзима Q10 и мемантина может несколько замедлять прогрессирование БГ. Важно с помощью нейролептиков и антидепрессантов корригировать сопутствующие психические расстройства, прежде всего депрессию, вспышки агрессии и неконтролируемого поведения [11].
При малой хорее средством выбора являются препараты вальпроевой кислоты и карбамазепины; только в том случае, если с их помощью не удается контролировать гиперкинез, назначают нейролептики в минимальной эффективной дозе. Кроме того, во избежание повторяющихся приступов ревматической лихорадки и развития порока сердца показана длительная пенициллинотерапия [9].
Тики представляют собой повторяющиеся отрывистые неритмичные движения, которые одномоментно вовлекают отдельные мышцы, группу мышц или часть тела. Тики возникают спонтанно на фоне нормальной двигательной активности и напоминают фрагменты целенаправленных движений. В отличие от многих других гиперкинезов больной может волевым усилием на определенное время (30–60 с) подавить тики, но обычно ценой быстро возрастающего внутреннего напряжения, которое неизбежно прорывается, вызывая кратковременную «бурю» тиков. Тикам может предшествовать ощущение непреодолимой потребности совершить движение, создающее иллюзию произвольности тика. Как правило, тики стереотипны и возникают в строго определенных у данного больного частях тела. Каждый больной имеет свой индивидуальный «репертуар» тиков, который меняется во времени. В отличие от других экстрапирамидных гиперкинезов тики сохраняются во сне [6].
Выделяют моторные, вокальные (фонические) и сенсорные тики, каждый из них, в свою очередь, делится на простые и сложные. К простым моторным тикам относят моргание, зажмуривание, подергивание головой, пожимание плечами, втягивание живота и др., к сложным — подпрыгивание, биение себя в грудь, эхопраксию (повторение жестов), копропраксию (воспроизведение неприличных жестов) и др. Простые моторные тики могут быть быстрыми, внезапными (клоническими) или более медленными и стойкими (дистоническими); например, к клоническим тикам относят моргание, а к дистоническим — зажмуривание (блефароспазм), окулогирные кризы, дистонические подергивания в области шеи, плеча, брюшных мышц. Простые вокальные тики включают покашливание, фырканье, похрюкивание, свист, сложные вокальные: эхолалию (повторение чужих слов); копролалию (произнесение непристойных слов); палилалию (повторение произнесенных самим больным слов или звуков). Сенсорные тики представляют собой кратковременные весьма неприятные ощущения, вынуждающие больного совершить движение. Они могут возникать в определенной части тела (например, в плече, кисти, животе или горле) и вынуждать больного совершать движение именно в этом регионе. По распространенности тики могут быть локальными (чаще в области лица, шеи, плечевого пояса), множественными или генерализованными.
Чаще всего тики имеют первичный характер, т. е. не связаны с каким-либо иным заболеванием, не сопровождаются другими двигательными синдромами, начинаются в детском и подростковом возрасте и обусловлены нарушением созревания связей между базальными ганглиями, лимбической системой и лобной корой. Мальчики страдают в 2–4 раза чаще, чем девочки [9].
Первичные тики условно подразделяют:
Есть основания полагать, что хронические моторные и вокальные тики и СТ, а возможно, и транзиторные тики могут быть проявлением одного и того же генетического дефекта, наследованного по аутосомно-доминантному типу. В то же время различия в выраженности гиперкинеза даже у однояйцевых близнецов указывают на важную роль внешних факторов, в частности действующих в перинатальном периоде [6].
Для СТ характерно волнообразное течение с периодами усиления и ослабления гиперкинеза, иногда с длительными спонтанными ремиссиями. Как правило, тики возникают у каждого больного в строго определенных частях тела. Каждый пациент имеет свой индивидуальный «репертуар» тиков, который меняется во времени. Обычно тики максимально выражены в подростковом периоде, а затем становятся слабее в юношеском и молодом возрасте. С наступлением зрелости примерно в трети случаев тики исчезают, у трети больных они значительно уменьшаются, а у оставшейся трети сохраняются в течение всей жизни, хотя и в этом случае редко приводят к инвалидизации. С возрастом уменьшается не только интенсивность тиков, но и их дезадаптирующее влияние. У большинства взрослых больных тики обычно не усиливаются, возможно лишь кратковременное ухудшение в период стрессовых ситуаций.
Более чем у половины больных с СТ выявляются сопутствующие психические расстройства (синдром навязчивых состояний, обсессивно-компульсивный синдром, синдром дефицита внимания с гиперактивностью), роль которых в социальной дезадаптации больного бывает подчас значительно выше, чем роль гиперкинеза.
Причиной вторичных тиков, которые встречаются реже, могут быть: повреждение головного мозга в перинатальном периоде, прием лекарственных препаратов (антиконвульсантов, нейролептиков, психостимуляторов и др.), черепно-мозговая травма, энцефалиты, сосудистые заболевания мозга, отравление угарным газом и т. д. При вторичных тиках гиперкинез обычно бывает менее динамичным (реже меняет локализацию, частоту, интенсивность), в меньшей степени оказываются выражены императивные позывы к движению и способность к подавлению гиперкинеза, могут присутствовать сопутствующие неврологические синдромы или такие психопатологические синдромы, как задержка психомоторного развития и умственная отсталость. Тиком иногда ошибочно называют доброкачественную миокимию век — преходящие подергивания круговой мышцы глаз, возникающие у вполне здоровых лиц при переутомлении, волнении, повышенном употреблении кофе или курении и не требующие лечения.
Во многих случаях при тиках медикаментозного лечения не требуется и достаточно успокоить больного и его родственников, рассказав о природе заболевания и указав на его доброкачественность. Важно отметить, что больному не угрожают снижение интеллекта, тяжелое психическое или неврологическое заболевание, и в подавляющем большинстве случаев такие пациенты хорошо социально адаптируются [9].
В легких случаях показано нефармакологическое воздействие в виде психопедагогической коррекции, обучения методам самоконтроля и саморегуляции. При умеренно выраженных тиках применяют бензодиазепины (клоназепам, 0,5–6 мг/сут) и другие ГАМКергические средства (баклофен, 20–75 мг/сут; фенибут, 250–1000 мг/сут). За рубежом для лечения умеренно выраженных тиков используют также клонидин и тетрабеназин. В более тяжелых случаях назначают «мягкие» нейролептики (сульпирид, 100–400 мг/сут; тиаприд, 200–400 мг/сут) либо атипичные нейролептики (например, рисперидон, 0,5–4 мг/сут или оланзапин, 2,5–5 мг/сут).
При резистентности к указанной терапии применяют большие дозы высокопотенциальных нейролептиков, комбинации двух нейролептиков с разным механизмом действия (например, рисперидона и тиаприда), комбинации нейролептика с антиконвульсантами (например, клоназепамом или топираматом) либо баклофеном. При тяжелых болезненных дистонических тиках, вовлекающих мышцы лица и шеи, возможно лечение ботулотоксином, который инъецируют в мышцы, участвующие в тике. Показан положительный эффект инъекций ботулотоксина в голосовые связки при вокальных тиках, в том числе при копролалии.
Для лечения сопутствующего синдрома нарушенного внимания и гиперактивности назначают ноотропные средства (пирацетам, пиридитол, глиатилин и др.), агонисты пресинаптических a2-адренорецепторов — клонидин и гуанфацин, малые дозы психостимуляторов, селегилин, трициклические антидепрессанты. Для лечения синдрома навязчивых состояний — антидепрессанты, ингибирующие обратный захват серотонина (кломипрамин, сертралин, флувоксамин и др.).
В лечении больных с тиками важное место принадлежит методам психотерапии. Они не способствуют уменьшению тиков, но, изменяя в благоприятную сторону отношение больных к тикам и корригируя сопутствующие психические нарушения, прежде всего синдром навязчивых состояний, улучшают социальную адаптацию больных. Обучение приемам релаксации позволяет больным снимать накапливающееся внутреннее напряжение. Разработаны специальные методики, тренирующие возможности больного произвольно контролировать тики (например, путем совершения конкурирующего движения при появлении ощущения, предваряющего тик).
Литература
О. С. Левин, доктор медицинских наук, профессор