Мозолистое тело, дисгенезия, атрофия, болезнь Маркиафавы-Биньями
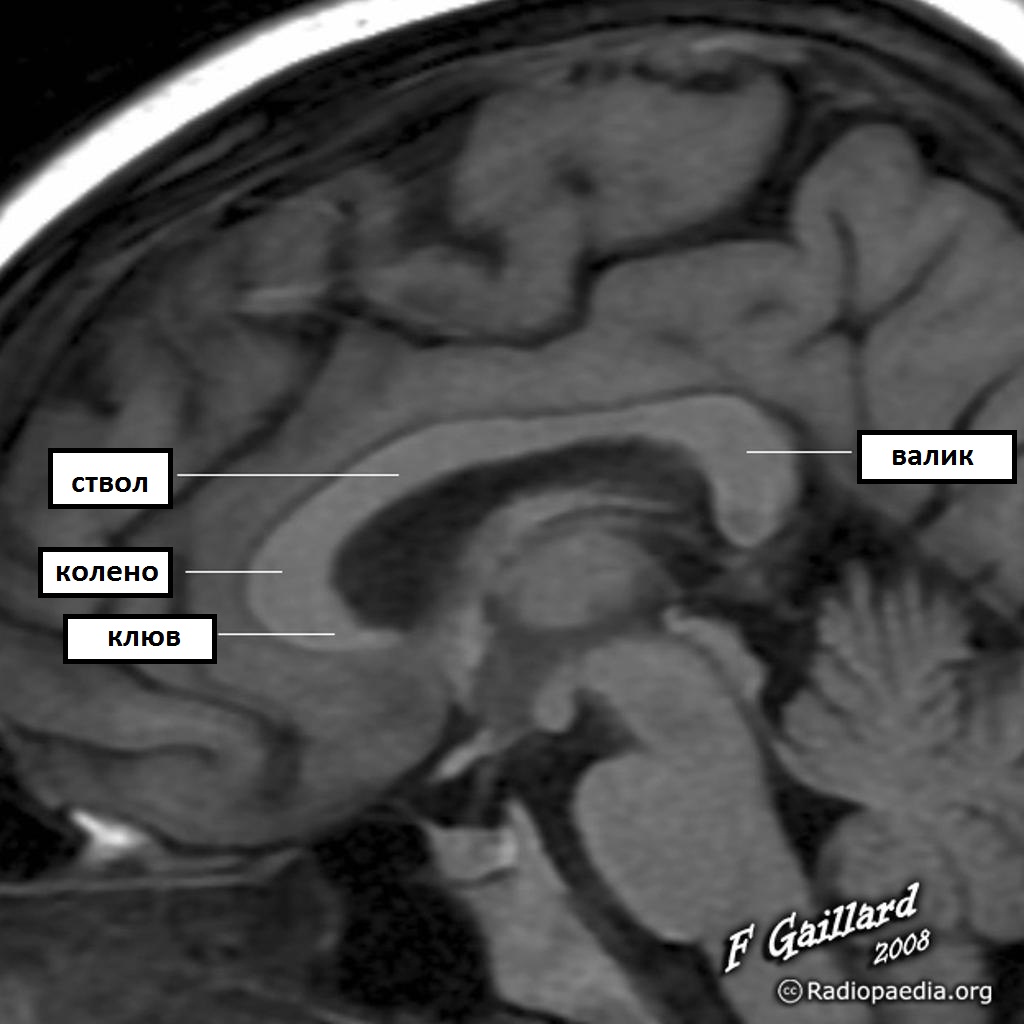
Мозолистое тело состоит:
Каждый отдел соединяет гомолатеральный отдел головного мозга.
Формирование мозолистого тела.
Мозолистое тело развивается в особом порядке:
От колена, затем тела, валик и в конце развивается клюв.
Миелинизация мозолистого тела идет от задних отделов к передним отделам.
Данные знания помогают сузить дифференциальный диагноз при патологиях мозолистого тела.
Дисгенезия vs Атрофия
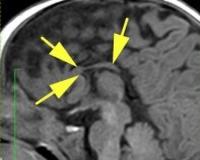
При дисгенезии мозолистого тела хорошо сформировано колено и передние отделы мозолистого тела, но отсутствует валик и клюв. Данная патология является врожденной. Патология представлена слева.
При атрофии мозолистого тела хорошо сформированы задние отделы мозолистого тела (задний отдел тела и валик), но при этом уменьшены в размерах клюв, колено и передний отдел тела. Данные изменения являются приобретенными.
Многие заболевания поражают мозолистое тело, поэтому наличие очагов не являются патогномоничным для определенного заболевания.

Болезнь Маркиафавы-Биньями (центральная дегенерация мозолистого тела, Маркиафавы синдром, экстрапонтинный миелинолиз).
Встречается у лиц злоупотребляющих алкоголем. У данных лиц на МРТ выявляется поражение валика и задних отделов ствола (тела) мозолистого тела.
На хронических стадиях болезни Маркиафавы-Биньями визуализируется мозолистое тело в виде сэндвича, при котором сохраняется верхних и нижних слоев мозолистого тела, но с некрозом средних слоев.
Агенезия мозолистого тела
Агенезия мозолистого тела — это врожденное отсутствие мозолистого тела либо его части. Аномалия обусловлена генетическими нарушениями, сосудистыми мальформациями, тератогенными факторами. Основные признаки заболевания: двигательные расстройства, задержка психоречевого развития, судорожные приступы. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначается церебральные КТ или МРТ, нейросонография у новорожденных, генетические исследования. Лечение симптоматическое: медикаментозная коррекция осложнений, реабилитационные программы.
МКБ-10
Общие сведения
Агенезия мозолистого тела (АМТ) — один из наиболее частых пороков нервной системы. Распространенность болезни в популяции составляет от 0,05% до 7% среди новорожденных, причем в группе детей с замедленным становлением психики агенезия встречается у 2,3%. Калифорнийская программа по изучению врожденных пороков предоставляет другие данные по частоте агенезии — 1,4 на 10000 живых новорожденных. Впервые состояние было описано в 1812 году в ходе аутопсии, проведенной немецким анатомом И. Рэйлем, и названо «природной моделью рассеченного мозга».
Причины
Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает мультифакториальная теория, согласно которой для формирования врожденного порока ЦНС требуется комбинация неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенезии:
Основным фактором риска выступает недоношенность. У новорожденных, родившихся до 27-недельного срока гестации МТ истончено в задних отделах, между 28 и 30 неделями — только в области валика. У рожденных после 30 недели в неонатальном периоде изменения не обнаруживаются, хотя при нейропсихологическом исследовании у школьников зачастую выявляется дефицит межполушарной передачи познавательной информации.
Патогенез
Мозолистое тело (МТ) представляет собой крупный пучок комиссуральных нервных волокон. Это важный аксональный путь, который соединяет соответствующие зоны коры правого и левого полушарий. Анатомическая структура имеет длину 7-9 см, состоит из более 300 млн. аксонов. Формирование МТ начинается на этапе позднего нейроонтогенеза (8-9 недели эмбриогенеза). Его созревание в норме продолжается до 20-25 лет.
Агенезия возникает при нарушении дифференциации нервной трубки в период со 2 до 5 месяца внутриутробного развития. При полном отсутствии МТ третий мозговой желудочек остается открытым, не формируются столбы свода мозга, отсутствуют прозрачные перегородки. В 60% случаев при АМТ передней комиссуры нет вообще. В 10% она увеличена и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах постнатального периода.
Характерным анатомическим изменением является колпоцефалия, при которой расширены задние отделы боковых церебральных желудочков. Состояние не относится к истинной гидроцефалии новорожденных, а обусловлено уменьшением кортикальных ассоциативных путей. Еще один типичный признак порока — пучки Пробста, представляющие собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
Классификация
В практической неврологии состояние подразделяют на тотальное, когда орган полностью отсутствует, и частичное (парциальное), при котором визуализационные методы не обнаруживают отдельные участки МТ. Это имеет решающее значение для тяжести клинической картины, возможных осложнений. В соответствии с патогенетическими особенностями формирования врожденных пороков, выделяют следующие 3 формы болезни:
Симптомы
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (при гипоплазии) до критических нервно-психических расстройств при его грубом недоразвитии, сопровождающемся другими врожденными пороками ЦНС. У новорожденных признаки патологии могут вовсе отсутствовать и проявляться по мере взросления младенца задержкой психомоторного развития.
Двигательные нарушения определяются у 35-40% пациентов. Они проявляются мышечной гипотонией или дистонией, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Дети позже начинают держать голову, испытывают затруднения при обучении сидению, ползанию, ходьбе. Могут отмечаться координационные нарушения, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги.
Мозолистое тело поддерживает связь между церебральными зонами, формирует межполушарную организацию высших психических процессов. При его агенезии либо гипоплазии у детей выявляются когнитивные расстройства. У новорожденных пациентов и в раннем детстве наблюдается задержка речи, снижение динамического компонента игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, расстройства памяти, при тотальной АМТ снижен коэффициент интеллекта.
Осложнения
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают мальформации кортикального развития (22,8%), межполушарные кисты (14,3%), голопрозэнцефалия (14,3%). К более редким сопутствующим аномалиям относят кисты и гипоплазию мозжечка, синдром Арнольда-Киари. До 20% новорожденных, кроме структур ЦНС, имеют пороки нескольких внутренних органов.
У 75% больных с тотальным поражением наблюдается симптоматическая эпилепсия височно-лобной локализации, в 66% случаев выражены когнитивные нарушения. У 16% пациентов формируются расстройства аутистического спектра. Изредка встречаются патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанной аномалии зрительных нервов.
Диагностика
В качестве первичного метода обследования в пренатальном периоде проводится акушерское УЗИ. У новорожденных для скрининговой диагностики используется нейросонография, однако этот метод не всегда показывает хорошую информативность, особенно при парциальной агенезии. Для верификации диагноза назначаются следующие методы исследования:
Лечение агенезии мозолистого тела
Специфическая терапия отсутствует. Медикаментозное лечение назначается неонатологом или педиатром индивидуально с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста используются антиконвульсанты, нейрометаболические препараты, дегидратационная терапия. Основу медицинской помощи составляет комплексная реабилитация, которая включает следующие составляющие:
Прогноз и профилактика
Прогноз определяется видом врожденной аномалии мозолистого тела, наличием сопутствующих пороков развития ЦНС. Благоприятный исход наблюдается при частичной гипоплазии МТ, а в случае комбинированных церебральных пороков у новорожденных могут быть жизнеугрожающие осложнения. Профилактические меры включают медико-генетическое консультирование, исключение тератогенных влияний в гестационном периоде.
Недоразвитие мозолистого тела (агенезия)
Развитие головного мозга ребенка начинается внутриутробно и активно продолжается после рождения.
По исследованиям физиологов правое полушарие головного мозга – гуманитарное, образное, творческое – отвечает за тело, координацию движений, баланс, пространственное зрительное и кинестетическое восприятие.
Левое полушарие головного мозга – математическое, знаковое, речевое, логическое, аналитическое – отвечает за восприятие – слуховой информации, постановку целей и построений программ.
Единство мозга складывается из деятельности двух полушарий, тесно связанных между собой системой нервных волокон (мозолистое тело).
Мозолистое тело (межполушарные связи) находится между полушариями головного мозга в теменно-затылочной части и состоит из двухсот миллионов нервных волокон. Оно необходимо для координации работы мозга и передачи информации из одного полушария в другое.
Агенезия (нарушение, недоразвитие) мозолистого тела искажает познавательную деятельность детей. Если нарушается проводимость через мозолистое тело, то ведущее полушарие берет на себя большую нагрузку, а другое блокируется. Оба полушарие начинают работать без связи.
Нарушаются пространственная ориентация, баланс, осознание собственного тела, адекватное эмоциональное реагирование, координация работы зрительного и аудиального восприятия с работой пишущей руки.
Ребенок с такими проблемами не ползает, тяжело начинает ходить, с большим трудом начинает читать и писать, воспринимая информацию на слух или зрительно. У детей с данной патологией, если вовремя не начать коррекцию и последующую реабилитацию, возникает целый ряд серьезных проблем, которые являются серьезным препятствием в развитии и обучении, в том числе и школьном.
В том случае, если агенезия мозолистого тела не сопровождается никакими другими патологиями развития, прогноз для больного достаточно благоприятный. Примерно восемьдесят с лишним процентов таких детей развиваются практически без нарушений либо с пограничными проблемами в неврологическом развитии. Нужно признать, что главная «опасность» этого нарушения таится в том, что у ребенка не происходит закрепления полученных умений и навыков навсегда, часто случаются «откаты», ребенок требует всё время поддерживающей терапии с нарастающей нагрузкой для мозга. Такой подход должен сохраняться до 12-14 лет ребенка, пока межполушарные связи окончательно не сформируются. Форсировать события здесь, к сожалению, нельзя. Иначе не избежать сочетанных проблем и других патологических состояний, которые усиливают симптоматику и ухудшают клиническую картину.
Агенезия мозолистого тела, хоть и встречается сравнительно часто, тем не менее, является малоизученным состоянием, особенно на просторах нашей страны.
Мозолистое тело истончено в головном мозге что это значит
Детский аутизм относится к нарушениям психического развития, протекающим с расстройством социальных и коммуникативных функций, снижением физической активности, со склонностью к двигательным и/или речевым стереотипиям.
Авторы статьи провели исследование с целью обобщения данных о клинико-диагностических особенностях и демонстрации редких наследственных синдромов, протекающих с клинической картиной детского аутизма, неврологическими расстройствами и структурно уменьшенным в размере МТ.
Диагностика детского аутизма включает анализ анамнестических данных, медико-генетические, неврологические, психопатологические, патопсихологические, лабораторные и инструментальные обследования. Ранняя диагностика ГМТ возможна на сроке беременности 18–20 нед. При подозрении на ГМТ у плода проводят пренатальное кариотипирование. На пренатальных диффузионно-тензорных МР-изображениях клюв и колено МТ практически сформированы к 15-й неделе гестации. Окончательное строение МТ приобретает к 20-й неделе гестации. На постнатальных МР-изображениях ГМТ визуализируется в виде равномерно истонченного контура с уменьшением его размера в переднезаднем направлении. Определенный интерес у больных с ГМТ представляет сочетание неврологических и психических симптомов.
Особенностью аутизма в раннем возрасте является неравномерное накопление лексического словаря; отсутствие подражательной функции и недостаточная коммуникативная направленность речи. У ряда пациентов отмечается общее недоразвитие речи; нарушения поведения; отсутствие реакции на собственное имя при анатомически неповрежденном слуховом анализаторе; гиперсенситивная реакция на свет, звуки и тактильные стимулы; могут наблюдаться эпилептические приступы, двигательные расстройства.
У пациентов старшего возраста, как правило, сохраняется интеллектуальная недостаточность вплоть до умственной отсталости; эпилептические приступы, входящие в структуру эпилепсии; слабая или практически отсутствующая дифференцированность эмоциональных проявлений; несформированность социально-коммуникативных навыков; недостаточное использование парадигматических средств общения (мимика, жесты); двигательные расстройства.
К настоящему времени установлено, что детский аутизм в сочетании с ГМТ ассоциирован с различными нейрогенетическими синдромами, характеризующимися неспецифичностью клинических проявлений, особенно на ранних стадиях болезни.
Специалисты проанализировали клинические случаи — у двух обследованных пациенток с нейрогенетическими синдромами, сопровождающимися на МРТ головного мозга гипоплазией МТ, были изучены психические и неврологические нарушения. Проведены комплексный анализ анамнестических данных; медико-генетическое, неврологическое, психопатологическое, патопсихологическое, лабораторные и инструментальные обследования.
У первой пациентки выявлен нейрогенетический синдром Вольфа-Хиршхорна, (46,XХ,t(4;6)(p16.3;p23),del(4)(p16.3)), у второй – синдром Прадера-Вилли, (46,XХ 15q11.2q13). В представленных двух наблюдениях отмечен отягощенный акушерский анамнез, неблагополучный анте и интранатальный периоды: угроза прерывания в I триместре, задержка внутриутробного развития плода в III триместре, преждевременные оперативные роды на сроке гестации 31 нед посредством экстренной операции кесарева сечения в связи с остро возникшей внутриутробной гипоксией плода в первом наблюдении; во втором: гестоз беременных в III триместре, экстренная операция кесарева сечения на сроке 34–35 нед. (преждевременная отслойка плаценты).
Обе пациентки имели низкую массу тела при рождении, признаки гипоксически-ишемического поражения головного мозга, более выраженные у первой пациентки (оценка по шкале Апгар 5/6 баллов, морфофункциональная незрелость, недоношенность 2-й степени), во втором случае – 7/8 баллов, недоношенность 1-й степени.
В дальнейшем низкие показатели оценки состояния анте-, интра- и постнатального периодов коррелировали с выраженностью психических, неврологических и когнитивных расстройств. К наиболее ярким системным клиническим проявлениям дизэмбриогенетических синдромов относятся психические и неврологические расстройства. В данной работе выраженность аутистических расстройств зависела от степени поражения головного мозга. В качестве иллюстрации тяжелого течения детского аутизма – первый клинический случай – больная девочка с тяжелой степенью умственной отсталости, тотальным недоразвитием всех уровней психической деятельности, отсутствием вербальных средств общения, двигательными стереотипиями, нарушением социального взаимодействия в сочетании с ранним органическим поражением головного мозга.
Второй случай – органический аутизм с умственной отсталостью легкой степени с негрубым нарушением социальной адаптации, недостаточной сформированностью отдельных структур 1-го и 2-го функционального блока мозга. Эпилептические проявления у первой пациентки представлены в виде структурной фокальной эпилепсии, подтвержденной результатами ЭЭГ (мультирегиональная эпилептиформная активность); у второй пациентки зарегистрирована непостоянная региональная эпилептиформная активность в лобно-височных отведениях слева, на момент осмотра, не требовавшая назначения противоэпилептической терапии. Важно отметить, что у первой больной выявлена тяжелая задержка психомоторного развития, предшествующая манифестации эпилептических приступов, что говорит о формировании выраженной церебральной патологии в дебюте заболевания. В первом случае на МРТ верифицирована гипоплазия задних отделов МТ, умеренные постгипоксические изменения вещества головного мозга; во втором – гипоплазия перешейка МТ, кортикальная субатрофия лобных долей, негрубое расширение наружных субарахноидальных пространств, ограниченные зоны перивентрикулярного глиоза.
У первой пациентки выявлен когнитивный дефицит, соответствующий тяжелой степени умственной отсталости; у второй пациентки – преобладали негрубые нарушения зрительно-пространственной координации, что коррелировало с целостностью задних отделов МТ. Становится понятным, что уровень когнитивного дефицита может быть в некоторой степени величиной, связанной с толщиной заднего отдела МТ. Это имеет свое объяснение: более высокая степень миелинизации нервных волокон способствует большей скорости распространения нервных импульсов по нервным волокнам, тем самым обеспечивая наилучшие результаты у обследуемых пациентов.
Таким образом, тяжесть клинической картины у больных девочек обусловлена наличием дизэмбриогенетических нарушений в сочетании с перинатальными осложнениями. В настоящее время специфическая терапия нейрогенетических синдромов не разработана. Тактика ведения подобных пациентов зависит от тяжести состояния, течения заболевания и включает в себя проведение регулярных реабилитационных мероприятий (массаж, лечебная физкультура), назначение симптоматической терапии, направленной на уменьшение выраженности проявлений спастичности. Важным компонентом лечения подобных больных является медико-психолого-педагогическое сопровождение, направленное на уменьшение выраженности аутистических расстройств.
Длительное воздействие неблагоприятных перинатальных факторов, способствующих нарушению нормального развития мозга, является одной из причин формирования ГМТ. Основное внимание в диагностике нейрогенетических синдромов принадлежит комплексному неврологическому, психиатрическому обследованию, нейропсихологическому тестированию, ЭЭГ-диагностике и нейровизуализационным техникам. Для окончательного подтверждения диагноза требуется ДНК-тестирование.
Динамика интеллектуальных расстройств при детском аутизме зависит от характера заболевания, в структуре которого наблюдается данное расстройство, одним из звеньев патогенеза которого является повреждение высших психических функций на этапах раннего онтогенеза. Относительно благоприятный прогноз заболевания отмечается при более позднем дебюте неврологического и психического дефицита. В структуру различных наследственных нейрогенетических синдромов входит сложная комбинация неврологических, психических, нейрофизиологических и радиологических нарушений; их своевременное распознавание требует большого клинического опыта, практических знаний не только в области неврологии, но и в смежных разделах медицины.
Анализ микроструктурных изменений мозолистого тела при помощи МР-трактографии у пациентов с эпилепсией
1 – ГУ «РНПЦ онкологии и медицинской радиологии им. Н.Н. Александрова» г. Минска, Беларусь
2 – ГУО «Белорусская медицинская академия последипломного образования» г. Минск, Беларусь.
В статье приведены результаты оценки МР-трактографии мозолистого тела в диагностике структурных повреждений головного мозга у пациентов с эпилепсией.
Ключевые слова: МР-трактография, эпилепсия, мозолистое тело.
Роль мозолистого тела (далее – МТ) в функционировании головного мозга сложно переоценить. Современные подходы к диагностике и технологическое развитие методов нейровизуализации открывает новые возможности в выявлении микроструктурных изменений головного мозга в целом и МТ в частности. По мнению ряда исследователей, оценка состояния МТ приобретает особое значение в анализе развития различных патологических состояний головного мозга.
МР-трактография, как методика оценки микроструктурных изменений, получила применение в диагностике аксональных повреждений, хронической ишемии головного мозга, болезни мотонейрона, острого диссеминированного энцефаломиелита, опухолей головного мозга и аномалиях развития ЦНС, кортикальных инфарктов, неопухолевых заболеваний (рассеянный склероз, болезнь Альцгеймера).
Несмотря на разнообразные клинические исследования, дифференцированные подходы к диагностике, использование различных дизайнов и протоколов нейровизуализации, в настоящее время нет четкого и однозначного понимания влияния различных отделов МТ на развитие и распространение эпилептической активности, вклад составных частей МТ в эпилептогенез, особенно при наличии сопутствующих патологических состояний головного мозга.
Кроме того, отсутствуют данные о взаимосвязи вовлечения МТ в патологический процесс с выраженностью клинических проявлений при опухолях, инсультах, воспалительных процессах, не уточнены критерии каллозальной атрофии из-за недостаточной определенности параметров главной мозговой спайки в популяции у здоровых лиц.
Многие исследователи отмечают, что диагностическая значимость МР-трактографии напрямую зависит от уровня компетенции исследователя (знания анатомии проводящих путей и умения соотнести их с МР-изображениями), а также может быть подвержена ошибкам, связанным с эффектом частичного объемного усреднения, шумом, некорректным выделением области интереса и т.п.
В рамках детального изучения микроструктурных изменений МТ современными методиками нейровизуализации (включая DTI), с учетом недостаточного количества и несистематизированности полученных сведений, требуются проведение оценки показателей FA и ADC МТ при различных патологических процессах, а также параметров транскаллезных и афферентных трактов МТ по данным МР-трактографии для сопоставления этих результатов с оценкой общего состояния проводящих путей головного мозга и определения прогнозов в отношении течения процесса, в частности – у пациентов с эпилепсией.
Показания к проведению МР-трактографии головного мозга:
Противопоказания к проведению МР-трактографии сводятся к противопоказаниям при проведении МРТ:
Ограничением при проведении МРТ является неадекватное состояние пациента и невозможность выполнения им требований к проведению МРТ. Дополнительной подготовки перед проведением МР-трактографии не требуется.
Цель исследования. Оценка данных рутинной МР-томографии и МР-трактографии в диагностике структурных повреждений головного мозга у пациентов с эпилепсией, выявление закономерностей изменения трактографической картины в зависимости от длительности приема противоэпилептических препаратов, тяжести течения заболевания. Показаниями к проведению МР-трактографии головного мозга в ходе исследования (критерии включения) считалось оценка поражений трактов при эпилепсии, измерение фракционной анизотропии (FA) и измеряемого коэффициента диффузии (ADC) для оценки микроструктурных изменений и определения дальнейшей тактики ведения пациента.
Материал и методы исследования. Нами обследовано 67 пациентов с эпилепсией, с использованием набора мультипланарных импульсных последовательностей и пакета диффузионной тензорной магнитно-резонансной томографии. Произведено сопоставление проведенных данных с контрольной группой (n=64). Средний возраст в группе пациентов с эпилепсией составил 29,1±3,2 лет.
Результаты и обсуждение. Структурные изменения при МРТ головного мозга у пациентов с эпилепсией были выявлены в 69,9% случаях: неокортикальные поражения у 21,9%, мезиальный темпоральный склероз – у 24,6%, их сочетание – у 30,1% обследованных; отсутствие визуальных морфологических изменений головного мозга было отмечено – у 23,4%. При этом у 71% пациентов было обнаружено расширение субарахноидальных пространств, в 24% случаев имела место их асимметрия с преобладанием патологических изменений на стороне очага, у 74% пациентов с эпилепсией наблюдалось расширение боковых желудочков, из них в 80% случаев – расширение височного рога бокового желудочка на стороне эпилептогенного очага.
Были получены следующие значения FA: для передних отделов мозга – 0,52 (0,5÷0,55), для задних – 0,53 (0,52÷0,54). Межполушарная асимметрия по данному параметру выявлена лишь для задних квадрантов, основная область которых представлена височными долями. Средняя диффузионная способность для передних отделов мозга составила 0,88 (0,86÷0,9), для задних – 0,89 (0,87÷0,91) без значимой асимметрии (р>0,05). Длина трактов в передних квадрантах составила 26,1 (23,8÷28,7) мм, в задних – 44,95 (41,5÷50,8) мм. При визуальной оценке симметричности трактов выявлено, что уменьшение их представленности на стороне очага характерно для пациентов с кортикальной дисплазией или экстрагиппокампальным расположением визуализируемых очагов поражения. В случае наличия мезиального темпорального склероза имело место «обеднение» трактографической картины в противоположном полушарии, что может объясняться феноменом гиппокампальной деафферентации. Наличие асимметрии трактов также было взаимосвязано со значениями FA и ADC – на стороне визуальных нарушений, где количество линий трактов (lines) было меньшим, отмечалось снижение фракционной анизотропии и увеличение средней диффузионной способности.
Было отмечено, что микроструктурные изменения в МТ коррелируют с когнитивной дисфункцией у пациентов с эпилепсией. Отмечается уменьшение количества афферентных трансколлозальных трактов у пациентов с выраженными когнитивными нарушениями. Для детальной оценки микроструктурных изменений в белом веществе с помощью DTI можно предположить формирующуюся потерю аксонов у пациентов с когнитивными нарушениями, что находит свое отражение в изменений трактографической картины. Когнитивные изменения коррелируют с DTI параметрами, отражая повреждение соединений МТ ведущее к ухудшению нервной проводимости. Таким образом, констатируется связь когнитивных нарушений с каллозальных повреждениями у пациентов с эпилепсией.
В ходе анализа в группе пациентов с эпилепсией отмечается снижение FA и повышение ADC МТ в сравнении с пациентами контрольной группы. При этом выявленное снижение FA МТ коррелировало с длительностью приема противоэпилептических препаратов (ПЭП). В литературе отмечается также снижение FA клюва и тела МТ, которое коррелирует с продолжительностью эпилепсии и отражает связь эпилепсии с микроструктурными изменения МТ. Полученные результаты позволяют предполагать, что ПЭП-терапия может привести к дополнительному повреждению и вторичной дегенерации МТ, напрямую связанных с эффектами лечения.
Заключение.
МР-трактография головного мозга у пациентов с эпилепсией позволяет получить детальную информацию о проводящих путях головного мозга, выявить их микроструктурные изменения, оценить выраженность сосудистых поражений, демиелинизации, глиоза (как сопутствующих состояний) на основе количественной оценки FA и ADC структур головного мозга, вовлекаемого в патологический процесс.
Комплексная оценка полученных результатов предполагает, что использование DTI в рутинной практике может с течением трансформироваться из «просто используемой», «рекомендуемой к применению» методики нейровизуализации в анализе микроструктурных изменений головного мозга и его структур, в «обязательно включенную» в пакет стандартных импульсных последовательностей, в особенности – в специализированных неврологических и нейрохирургических стационарах. Необходима более детальная оценка вклада DTI в понимание характера патоморфологических изменений проводящих путей головного мозга на макро- и микроструктурном уровне при различных патологических процессах и выработка оптимального алгоритма интерпретации полученных данных.