Митохондриальная ДНК


Митохондриальная ДНК (мтДНК) — ДНК, находящаяся (в отличие от ядерной ДНК) в митохондриях, органоидах эукариотических клеток.
Содержание
История открытия
Митохондриальная ДНК была открыта Маргит Насс и Сильвен Насс в 1963 году в Стокгольмском университете при помощи электронной микроскопии [2] и, независимо, учёными Эллен Харлсбруннер, Хансом Туппи и Готтфридом Шацем при биохимическом анализе фракций митохондрий дрожжей в Венском университете в 1964 году. [3]
Теории возникновения митохондриальной ДНК
На основании сходства в последовательностях нуклеотидов ДНК ближайшими родственниками митохондрий среди ныне живущих прокариот считают альфа-протеобактерий (выдвигалась также гипотеза, что к митохондриям близки риккетсии). Сравнительный анализ геномов митохондрий показывает, что в ходе эволюции происходило постепенное перемещение генов предков современных митохондрий в ядро клетки. Необъяснимыми с эволюционной точки зрения остаются некоторые особенности митохондриальной ДНК (например, довольно большое число интронов, нетрадиционное использование триплетов и др.). Ввиду ограниченного размера митохондриального генома бо́льшая часть митохондриальных белков кодируется в ядре. При этом бо́льшая часть митохондриальных тРНК кодируются митохондриальным геномом.
Формы и число молекул митохондриальной ДНК

У большинства изученных организмов митохондрии содержат только кольцевые молекулы ДНК, у некоторых растений одновременно присутствуют и кольцевые, и линейные молекулы, а у ряда протистов (например, инфузорий) имеются только линейные молекулы. [5]
Митохондрии млекопитающих обычно содержат от двух до десяти идентичных копий кольцевых молекул ДНК. [6]
У растений каждая митохондрия содержит несколько молекул ДНК разного размера, которые способны к рекомбинации.
У протистов из отряда кинетопластид (например, у трипаносом) в особом участке митохондрии (кинетопласте) содержится два типа молекул ДНК — идентичные макси-кольца (20-50 штук) длиной около 21 т.п.о. и мини-кольца (20 000 — 55 000 штук, около 300 разновидностей, средняя длина около 1000 п.о.). Все кольца соединены в единую сеть (катенаны), которая разрушается и восстанавливается при каждом цикле репликации. Макси-кольца гомологичны митохондриальной ДНК других организмов. Каждое мини-кольцо содержит четыре сходных консервативных участка и четыре уникальных гипервариабельных участка. [7] В мини-кольцах закодированы короткие молекулы направляющих РНК (guideRNA), которые осуществляют редактирование РНК, транскрибируемых с генов макси-колец.
Устойчивость митохондриальной ДНК
Митохондриальная ДНК особенно чувствительна к активным формам кислорода, генерируемым дыхательной цепью, в связи с непосредственной их близостью. Хотя митохондриальная ДНК связана с белками, их защитная роль менее выражена, чем в случае ядерной ДНК. Мутации в ДНК митохондрий могут вызывать передаваемые по материнской линии наследственные заболевания. Также имеются данные, указывающие на возможный вклад мутаций митохондриальной ДНК в процесс старения и развитие возрастных патологий. [8] У человека митохондриальная ДНК обычно присутствует в количестве 100—10000 копий на клетку (сперматозоиды и яйцеклетки являются исключением). С множественностью митохондриальных геномов связаны особенности проявления митохондриальных болезней — обычно позднее их начало и очень изменчивые симптомы.
Митохондриальная наследственность
Наследование по материнской линии
У большинства многоклеточных организмов митохондриальная ДНК наследуется по материнской линии. Яйцеклетка содержит на несколько порядков больше копий митохондриальной ДНК, чем сперматозоид. В сперматозоиде обычно не больше десятка митохондрий (у человека — одна спирально закрученная митохондрия), в небольших яйцеклетках морского ежа — несколько сотен тысяч, а в крупных ооцитах лягушки — десятки миллионов. Кроме того, обычно происходит деградация митохондрий сперматозоида после оплодотворения. [9]
При половом размножении митохондрии, как правило, наследуются исключительно по материнской линии, митохондрии сперматозоида обычно разрушаются после оплодотворения. Кроме того, большая часть митохондрий сперматозоида находятся в основании жгутика, которое при оплодотворении иногда теряется. В 1999 году было обнаружено, что митохондрии сперматозоидов помечены убиквитином (белком-меткой, которая приводит к разрушению отцовских митохондрий в зиготе). [10]
Так как митохондриальная ДНК не является высококонсервативной и имеет высокую скорость мутирования, она является хорошим объектом для изучения филогении (эволюционного родства) живых организмов. Для этого определяют последовательности митохондриальной ДНК у разных видов и сравнивают их при помощи специальных компьютерных программ и получают эволюционное древо для изученных видов. Исследование митохондриальных ДНК собак позволило проследить происхождение собак от диких волков. [11] Исследование митохондриальной ДНК в популяциях человека позволило вычислить «митохондриальную Еву», гипотетическую прародительницу всех живущих в настоящее время людей.
Наследование по отцовской линии
Геном митохондрий
Один из наиболее маленьких митохондриальных геномов имеет малярийный плазмодий (около 6.000 п.о., содержит два гена рРНК и три гена, кодирующих белки).
Недавно открытые рудиментарные митохондрии (митосомы) некоторых протистов (дизентерийной амёбы, микроспоридий и лямблий) не содержат ДНК. [25]
Некоторые растения имеют огромные молекулы митохондриальной ДНК (до 25 миллионов пар оснований), при этом содержащие примерно те же гены и в том же количестве, что и меньшие мтДНК. Длина митохондриальной ДНК может широко варьировать даже у растений одного семейства. В митохондриальной ДНК растений имеются некодирующие повторяющиеся последовательности.
Геном митохондрий человека кодирует следующие белки и РНК:
Особенности митохондриальной ДНК
Применение
Кроме изучения для построения различных филогенетических теорий, изучение митохондриального генома — основной инструмент при проведении идентификации. Возможность идентификации связана с существующими в митохондриальном геноме человека групповыми и даже индивидуальными различиями.
Наука
In the coming weeks, this wiki’s URL will be migrated to the primary fandom.com domain. Read more here
Митохондриальная ДНК
Схема митохондриального генома человека
Митохондриальная ДНК (мтДНК) — ДНК, находящаяся (в отличие от ядерной ДНК) в митохондриях, органоидах эукариотических клеток.
Содержание
История открытия
Теории возникновения митохондриальной ДНК
На основании сходства в последовательностях нуклеотидов ДНК ближайшими родственниками митохондрий среди ныне живущих прокариот считают альфа-протеобактерий (в частности, выдвигалась гипотеза, что к митохондриям близки риккетсии ). Сравнительный анализ геномов митохондрий показывает, что в ходе эволюции происходило постепенное перемещение генов предков современных митохондрий в ядро клетки. Необъяснимыми с эволюционной точки зрения остаются некоторые особенности митохондриальной ДНК (например, довольно большое число интронов, нетрадиционное использование триплетов и другие). Ввиду ограниченного размера митохондриального генома бо́льшая часть митохондриальных белков кодируется в ядре. При этом бо́льшая часть митохондриальных тРНК кодируются митохондриальным геномом.
Формы и число молекул митохондриальной ДНК
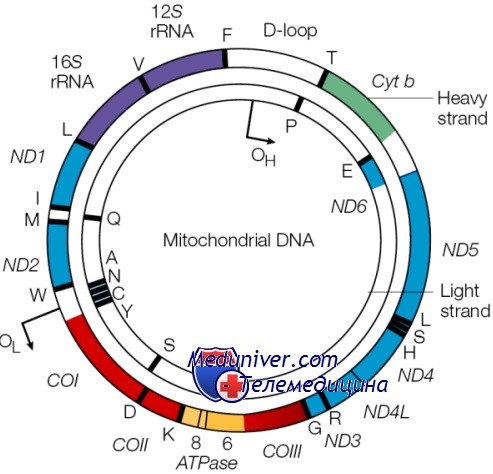
Электронная микроскопия демонстрирует определённую локализацию мтДНК в митохондриях человека. Разрешение 200 нм. (A) Сечение через цитоплазму после окрашивания мтДНК частичками золота. (B) Цитоплазма после экстракции; мтДНК, связанные с частичками золота, остались на месте. Из статьи Iborra et al., 2004. [4]
У большинства изученных организмов митохондрии содержат только кольцевые молекулы ДНК, у некоторых растений одновременно присутствуют и кольцевые, и линейные молекулы, а у ряда протистов (например, инфузорий ) имеются только линейные молекулы. [5]
Митохондрии млекопитающих обычно содержат от двух до десяти идентичных копий кольцевых молекул ДНК. [6]
У растений каждая митохондрия содержит несколько молекул ДНК разного размера, которые способны к рекомбинации.
У протистов из отряда кинетопластид (например, у трипаносом ) в особом участке митохондрии ( кинетопласте ) содержится два типа молекул ДНК — идентичные макси-кольца (20-50 штук) длиной около 21 т.п.о. и мини-кольца (20 000 — 55 000 штук, около 300 разновидностей, средняя длина около 1000 п.о.). Все кольца соединены в единую сеть ( катенаны ), которая разрушается и восстанавливается при каждом цикле репликации. Макси-кольца гомологичны митохондриальной ДНК других организмов. Каждое мини-кольцо содержит четыре сходных консервативных участка и четыре уникальных гипервариабельных участка. [7] В мини-кольцах закодированы короткие молекулы направляющих РНК ( guideRNA ), которые осуществляют редактирование РНК, транскрибируемых с генов макси-колец.
Устойчивость митохондриальной ДНК
Митохондриальная наследственность
Наследование по материнской линии
У большинства многоклеточных организмов митохондриальная ДНК наследуется по материнской линии. Яйцеклетка содержит на несколько порядков больше копий митохондриальной ДНК, чем сперматозоид. В сперматозоиде обычно не больше десятка митохондрий (у человека — одна спирально закрученная митохондрия), в небольших яйцеклетках морского ежа — несколько сотен тысяч, а в крупных ооцитах лягушки — десятки миллионов. Кроме того, обычно происходит деградация митохондрий сперматозоида после оплодотворения. [9]
Так как митохондриальная ДНК не является высококонсервативной и имеет высокую скорость мутирования, она является хорошим объектом для изучения филогении (эволюционного родства) живых организмов. Для этого определяют последовательности митохондриальной ДНК у разных видов и сравнивают их при помощи специальных компьютерных программ и получают эволюционное древо для изученных видов. Исследование митохондриальных ДНК собак позволило проследить происхождение собак от диких волков. [11] Исследование митохондриальной ДНК в популяциях человека позволило вычислить « митохондриальную Еву », гипотетическую прародительницу всех живущих в настоящее время людей.
Наследование по отцовской линии
Геном митохондрий
Один из наиболее маленьких митохондриальных геномов имеет малярийный плазмодий (около 6.000 п.о., содержит два гена рРНК и три гена, кодирующих белки).
Некоторые растения имеют огромные молекулы митохондриальной ДНК (до 25 миллионов пар оснований), при этом содержащие примерно те же гены и в том же количестве, что и меньшие мтДНК. Длина митохондриальной ДНК может широко варьировать даже у растений одного семейства. В митохондриальной ДНК растений имеются некодирующие повторяющиеся последовательности.
Геном митохондрий человека кодирует следующие белки и РНК:
Особенности митохондриальной ДНК
Применение
Примечания
Ссылки
См. также
Пурины ( Аденин • Гуанин ) • Пиримидины (Урацил • Тимин • Цитозин )
Митохондрии способны так как содержат днк
Некоторые родословные наследственных болезней не могут объясняться типичным менделирующим наследованием ядерных генов. Теперь известно, что они вызваны мутациями митохондриального генома и проявляют материнское наследование. Болезни, вызываемые мутациями в митДНК, демонстрируют множество необычных особенностей, происходящих из уникальных характеристик биологии и функции митохондрий.
Митохондриальный геном
Не вся РНК и белок, синтезируемые в клетке, кодируются ДНК ядра; небольшая, но важная доля кодируется в генах митохондриального генома. Этот геном состоит из кольцевой хромосомы размером 16,5 килобазы, располагающейся в органеллах митохондриях, а не в ядре. Большинство клеток содержит по крайней мере 1000 молекул митДНК, распределенных по сотням отдельных митохондрий. Важное исключение — зрелый овоцит, имеющий более 100 000 копий митДНК, формирующих до одной трети общего содержания ДНК в этих клетках.
Митохондриальная хромосома содержит 37 генов. Они кодируют 13 полипептидов — компонентов ферментов окислительного фосфорилирования, два типа рРНК и 22 тРНК, необходимых для трансляции транскриптов генов митохондрий. Остальные полипептиды комплекса окислительного фосфорилирования кодируются ядерным геномом.
В митДНК обнаружено более 100 различных перестроек и 100 разных точковых мутаций, вызывающих болезни у человека, часто поражающие ЦНС и мышечно-скелетную систему (например, миоклонус-эпилепсия с «рваными» красными волокнами — MERRF). Болезни, вызванные этими мутациями, имеют отличающийся тип наследования из-за трех необычных характеристик митохондрий: репликативной сегрегации, гомоплазмии и гетероплазмии, а также материнского наследования.
Репликативная сегрегация митохондриальной хромосомы
Первая уникальная характеристика митохондриальной хромосомы — отсутствие управляемой сегрегации, наблюдаемой в митозе и мейозе 46 ядерных хромосом. При делении клетки многочисленные копии митДНК в каждой митохондрии клетки копируются и произвольно расходятся во вновь синтезированные митохондрии. Митохондрии, в свою очередь, случайно распределяются между дочерними клетками. Этот процесс известен как репликативная сегрегация.
Гомоплазмия и гетероплазмия митохондриальной хромосомы
Вторая уникальная характеристика генетики митДНК возникает из-за того, что большинство клеток содержат много копий молекул митДНК. Когда мутация возникает в митДНК, она сначала присутствует только в одной из молекул в митохондрии. В ходе репликативной сегрегации митохондрия, содержащая мутантную митДНК, производит многочисленные копии мутантнои молекулы.
При делении клетка, содержащая смесь нормальных и мутантных митохондриальных ДНК, может передавать в дочерние клетки весьма различающиеся пропорции мутантнои и дикой митДНК. Одна дочерняя клетка может случайно получить митохондрии, содержащие чистую популяцию нормальных или чистую популяцию мутантных митохондриальных ДНК (ситуация, известная как гомоплазмия). Кроме того, дочерняя клетка может получить смесь митохондрий с мутацией и без нее (гетероплазмия).
Поскольку фенотипическая экспрессия мутации в митДНК зависит от относительных пропорций нормальной и мутантнои митДНК в клетках, формирующих различные ткани, неполная пенетрантность, переменная экспрессивность и плейотропия — типичные характеристики митохондриальных болезней.
Материнское наследование митохондриальной ДНК
Результат, определенный характеристиками генетики митДНК, называется материнским наследованием. Митохондрии сперматозоидов обычно отсутствуют в эмбрионе, поэтому митДНК наследуется от матери. Таким образом, все дети женщины, гомоплазмической по мутации митДНК, унаследуют мутацию, тогда как ни один из потомства мужчины, несущего ту же мутацию, не унаследует дефектную ДНК.
Материнское наследование гомоплазмической мутации митДНК, вызывающей наследственную нейропатию зрительного нерва Лебера.
Особенности материнского наследования при гетероплазмии у матери выявляют дополнительные характеристики генетики митДНК, имеющие медицинское значение. Во-первых, небольшое число молекул митДНК в развивающихся овоцитах впоследствии увеличивается до огромного количества, наблюдаемого в зрелых овоцитах. Это ограничение с последующим умножением митДНК в ходе овогенеза характеризуют так называемое «бутылочное горлышко» генетики митохондрий.
Именно поэтому вариабельность процентного содержания мутантных молекул митДНК, обнаруживаемая в потомстве матери с гетероплазмией, возникает, по крайней мере частично, вследствие увеличения только части митохондриальных хромосом в овогенезе. Можно ожидать, что мать с высокой пропорцией мутантных молекул митДНК более вероятно произведет яйцеклетки с высокой пропорцией мутантных молекул митДНК, и, следовательно, более клинически пораженное потомство, чем мать с более низкой пропорцией. Есть одно исключение из материнского наследования, когда у матери имеется гетероплазмия по делеции в митДНК; по неизвестным причинам делеционная молекула митДНК обычно не передается от клинически больных матерей их детям.
Хотя митохондрии почти всегда наследуются исключительно через мать, существует, по крайней мере, один пример отцовского наследования митДНК у пациентов с митохондриальной миопатией. Следовательно, у пациентов с наблюдаемыми спорадическими мутациями митДНК должна учитываться редкая возможность отцовского наследования митДНК.
Точное определение семейной родословной — важная часть работы с каждым пациентом. Родословные могут демонстрировать как типичные менделирующие варианты наследования, так и более редкие, вызванные митохондриальными мутациями и половым мозаицизмом; или сложные варианты семейных случаев, не соответствующие ни одному из типов наследования. Определение типа наследования важно не только для установления диагноза у пробанда, это также идентифицирует других индивидуумов в семье, находящихся в группе риска и нуждающихся в обследовании и консультировании.
Несмотря на сложные цитогенетические и молекулярные анализы, используемые генетиками, точная семейная история, включая родословную семьи, остается фундаментальным средством для всех врачей и генетических консультантов, используемым при планировании индивидуального лечения пациентов.
Характеристика митохондриального наследования:
• Женщины, гомоплазматические по мутации, передают эту мутацию всем детям; мужчины с аналогичной мутацией — нет.
• Женщины, гетероплазматические по точко-вым мутациям и дупликациям, передают их всем детям. Доля мутантных митохондрий у потомков и, следовательно, риск развития и тяжесть болезни могут значительно меняться в зависимости от доли мутантных митохондрий у матери, а также случайно, вследствие небольшого числа митохондрий в «бутылочном горлышке» при созревании овоцитов. Гетероплазматические делеции обычно не наследуются.
• Доля мутантных митохондрий в разных тканях гетероплазматических по мутации пациентов может значительно меняться, вызывая различные проявления болезни в одной семье с гетероплазмией митохондриальной мутации. Часто наблюдаются плейотропизм и вариабельная экспрессивность у разных больных в одной семье.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Митохондриальная патология у детей
Представлены данные о митохондриальных болезнях в детском возрасте. Рассматриваются исторические, этиопатогенетические и генетические аспекты этой группы болезней. Описаны основные виды митохондриальной патологии, подходы к диагностике и современные предс
The article focuses on mitochondrial disorders in infancy and childhood. Authors consider historical, etiopathogenic and genetic aspects of this group of diseases. Main types of mitochondrial pathologies are described, as well as approaches to diagnostics and contemporary ideas of potential treatment methods.
Митохондриальные болезни — это группа наследственной патологии, возникающей в результате нарушений клеточной энергетики, характеризующаяся полиморфизмом клинических проявлений, выражающаяся в преимущественном поражении центральной нервной системы и мышечной системы, а также других органов и систем организма [1].
Альтернативное определение митохондриальной патологии гласит, что это обширная группа патологических состояний, обусловленных генетическими, структурными и биохимическими дефектами митохондрий, нарушением тканевого дыхания и, как следствие, недостаточностью энергетического обмена.
Как указывает A. Munnich, «митохондриальные заболевания могут вызывать любой симптом, в любой ткани, в любом возрасте, при любом типе наследования» [2].
Митохондриальные дыхательные цепи — главный конечный путь аэробного метаболизма. Поэтому митохондриальную патологию нередко называют «болезнями дыхательной цепи митохондрий» (БДЦМ); это сравнительно новый класс болезней.
Исторические аспекты митохондриальной патологии
R. Luft и соавт. (1962) обнаружили взаимосвязь между мышечной слабостью и нарушениями процессов окислительного фосфорилирования в мышечной ткани [3]. S. Nass и M. Nass (1963) открыли существование собственного генетического аппарата митохондрий (обнаружены несколько копий кольцевой хромосомы) [4, 5]. В 1960–1970 гг. появилась концепция митохондриальных болезней, то есть патологии, этиологически опосредованной митохондриальной дисфункцией. В 1980-е гг. были получены точные молекулярно-генетические доказательства митохондриальной природы ряда заболеваний (болезнь Лебера, синдром Пирсона) [6].
Этиопатогенетические аспекты митохондриальной патологии
В зависимости от наличия основного метаболического дефекта принято рассматривать четыре основных группы митохондриальных болезней: 1) нарушения обмена пирувата; 2) дефекты обмена жирных кислот; 3) нарушения цикла Кребса; 4) дефекты электронного транспорта и окислительного фосфорилирования (OXPHOS) [1, 2].
Причинами возникновения митохондриальной патологии являются мутации в генах, кодирующих белки, задействованные в процессах энергообмена в клетках (включая субъединицы комплекса пируватдегидрогеназы, ферменты цикла Кребса, компоненты цепи транспорта электронов, структурные белки цепи транспорта электронов (ЦТЭ), митохондриальные транспортеры внутренней мембраны, регуляторы митохондриального нуклеотидного пула, а также факторы, взаимодействующие с ДНК митохондрий (мтДНК) [2, 6].
Митохондриальные нарушения связаны с большим числом болезней, не являющихся первичными митохондриальными цитопатиями. Тем не менее, при этих болезнях нарушения функций митохондрий вносят значимый вклад в патогенез и клинические проявления заболеваний. Описываемые болезни могут быть метаболическими, дегенеративными, воспалительными, врожденными/приобретенными мальформациями, а также неоплазмами.
Митохондрия является органеллой, которая присутствует практически в каждой клетке, за исключением зрелых эритроцитов. Именно поэтому митохондриальные болезни могут поражать любые системы и органы человеческого организма [6]. В связи с этим правильнее называть эти состояния «митохондриальными цитопатиями» [1, 2].
Основные особенности митохондриальных цитопатий включают выраженный полиморфизм клинических симптомов, мультисистемный характер поражения, вариабельность течения, прогрессирование и неадекватное реагирование на применяемую терапию.
Конечным результатом окислительного фосфорилирования, происходящего в комплексах 1-γ, является производство энергии (АТФ). Аденозин трифосфат — основной источник энергии для клеток.
Митохондриальная ДНК тесно взаимодействует с ядерной ДНК (яДНК). В каждом из 5 дыхательных комплексов основная часть субъединиц кодируется яДНК, а не мтДНК. Комплекс I состоит из 41 субъединицы, из которых 7 кодируются мтДНК, а остальные — яДНК. Комплекс II имеет всего 4 субъединицы; большая их часть кодируется яДНК. Комплекс III представлен десятью субъединицами; кодирование мтДНК — 1, яДНК — 9. Комплекс IV имеет 13 субъединиц, из которых 3 кодируются мтДНК, а 10 — яДНК. Комплекс V включает 12 субъединиц, кодирование мтДНК — 2, яДНК — 10 [2, 6].
Нарушения клеточной энергетики приводят к полисистемным заболеваниям. В первую очередь, страдают органы и ткани, являющиеся наиболее энергозависимыми: нервная система (энцефалопатии, полинейропатии), мышечная система (миопатии), сердце (кардиомиопатии), почки, печень, эндокринная система и другие органы и системы. До недавнего времени все эти заболевания определялись под многочисленными масками других нозологических форм патологии. К настоящему времени выявлено более 200 заболеваний, причиной которых являются мутации митохондриальной ДНК [1, 2, 6].
Митохондриальные болезни могут быть обусловлены патологией как митохондриального, так и ядерного генома. Как указывают P. F. Chinnery и соавт. (2004) и S. DiMauro (2004), мутации мтДНК были выявлены в 1 случае на 8000 населения, а распространенность митохондриальных заболеваний составляет порядка 11,5 случаев на 100 тысяч населения [7, 8].
В каждой клетке находятся от нескольких сотен до нескольких тысяч органелл — митохондрий, содержащих от 2 до 10 кольцевых молекул митохондриальной ДНК, способных к репликации, транскрипции и трансляции, причем независимо от ядерной ДНК.
Генетические аспекты митохондриальной патологии
Митохондриальная генетика отличается от классической менделевской в трех важнейших аспектах: 1) материнское наследование (всю цитоплазму, вместе с находящимися в ней органеллами, потомки получают вместе с яйцеклеткой); 2) гетероплазмия — одновременное существование в клетке нормального (дикого) и мутантного типов ДНК; 3) митотическая сегрегация (оба типа мтДНК в процессе деления клетки могут распределяться случайным образом между дочерними клетками) [1, 2].
Митохондриальная ДНК накапливает мутации более чем в 10 раз быстрее ядерного генома, так как она лишена защитных гистонов и ее окружение чрезвычайно богато реактивными видами кислорода, являющимися побочным продуктом метаболических процессов, протекающих в митохондриях. Пропорция мутантной мтДНК должна превышать критический пороговый уровень, прежде чем клетки начнут проявлять биохимические аномалии митохондриальных дыхательных цепей (пороговый эффект). Процентный уровень мутантной мтДНК может варьировать у индивидов внутри семей, а также в органах и тканях. В этом заключается одно из объяснений вариабельности клинической картины у больных с митохондриальными дисфункциями. Одни и те же мутации могут вызывать различные клинические синдромы (например, мутация A3243G — энцефалопатию с инсультоподобными пароксизмами — синдром MELAS, а также хроническую прогрессирующую наружную офтальмоплегию, сахарный диабет). Мутации в различных генах могут быть причиной одного и того же синдрома. Классическим примером такой ситуации является синдром MELAS [2].
Разновидности митохондриальной патологии
Если перечислить основные митохондриальные болезни, то в их числе окажутся следующие: митохондриальная нейрогастроинтестинальная энцефалопатия (MNGIE), синдром множественных делеций митохондриальной ДНК, липидная миопатия с нормальными уровнями карнитина, недостаточность карнитин пальмитоилтрансферазы, митохондриальный сахарный диабет, болезнь Альперса–Хуттенлохера, синдром Кернса–Сейра, болезнь Лебера (LHON), синдром Вольфрама, синдром MEMSA, синдром Пирсона, синдром SANDO, синдром MIRAS, синдром MELAS, синдром MERRF, синдром SCAE, синдром NARP, синдром Барта, синдром CPEO, синдром Ли и др. [1].
Наиболее часто в детском возрасте встречаются следующие клинические синдромы митохондриальной патологии: синдром MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз и инсультоподобные пароксизмы), синдром MERRF (миоклонус-эпилепсия с рваными красными волокнами), синдром Кернса–Сейра (характеризуется птозом, офтальмоплегией, пигментным ретинитом, атаксией, нарушением сердечного проведения), синдром NARP (нейропатия, атаксия, пигментный ретинит), синдром Ли (подострая некротизирующая энцефаломиелопатия), болезнь Лебера (наследственная оптическая нейропатия) [1, 2].
Имеется большой пул заболеваний, причиной которых является не мутации митохондриальной ДНК, а мутации ядерной ДНК, кодирующей работу митохондрий. К ним относятся следующие виды патологии: болезнь Барта (миопатия, кардиомиопатия, транзиторные нейтро- и тромбоцитопении), митохондриальная гастроинтестинальная энцефалопатия (аутосомно-рецессивное мультисистемное заболевание): птоз, офтальмоплегия, периферическая нейропатия, гастроинтестинальная дисфункция, приводящая к кахексии, лейкоэнцефалопатия. Возраст дебюта последнего заболевания весьма вариабелен — от периода новорожденности до 43 лет.
Диагностика митохондриальной патологии
Клинические критерии диагностики митохондриальных болезней сравнительно многочисленны: 1) миопатический симптомокомплекс (непереносимость физических нагрузок, мышечная слабость, снижение мышечного тонуса); 2) судороги (миоклонические или мультифокальные); 3) мозжечковый синдром (атаксия, интенционный тремор); 4) поражение глазодвигательных нервов (птоз, наружная офтальмоплегия); 5) полинейропатия; 6) инсультоподобные пароксизмы; 7) мигренеподобные головные боли; 8) черепно-лицевая дисморфия; 9) дисметаболические проявления (рвота, эпизоды летаргии, комы); 10) дыхательные нарушения (апноэ, гипервентиляция, тахипноэ); 11) поражение сердца, печени, почек; 12) прогрессирующее течение заболевания [1, 2].
В диагностике митохондриальных болезней используются следующие клинические критерии: 1) признаки поражения соединительной ткани (гипермобильный синдром, гиперэластичность кожи, нарушения осанки и др.); 2) нейродегенеративные проявления, лейкопатии при проведении магнитно-резонансной томографии (МРТ) головного мозга; 3) повторные эпизоды нарушения сознания или необъяснимые эпизоды рвоты у новорожденных; 4) необъяснимая атаксия; 5) отставание в умственном развитии без определенных причин; 6) отягощенный семейный анамнез; 7) внезапное ухудшение состояния ребенка (судороги, рвота, расстройства дыхания, вялость, слабость, нарушения мышечного тонуса — чаще мышечная гипотония, кома, летаргия; поражение печени и почек, не поддающееся обычной терапии) [1, 2].
Лабораторные (биохимические) исследования нацелены в первую очередь на выявление у пациентов лактат-ацидоза и/или пируват-ацидоза. При этом следует помнить, что нормальные показатели молочной кислоты не исключают наличия митохондриального заболевания. Другие биохимические показатели, исследуемые при подозрении на наличие митохондриальной патологии, включают кетоновые тела в крови и моче, ацилкарнитины плазмы крови, а также содержание органических кислот и аминокислот в крови и моче [9].
M. V. Miles и соавт. (2008) предложили оценивать содержание мышечного коэнзима Q10 у детей с дефектом ферментов дыхательной цепи митохондрий [10].
Цитоморфоденситометрические исследования позволяют оценивать активность митохондрий лимфоцитов (снижение количества, увеличение объема, снижение активности).
Из инструментальных исследований (помимо методов нейровизуализации) используется биопсия скелетных мышц с проведением специфических гистохимических реакций — для выявления феномена «рваных красных волокон» (ragged red fibers — RRF) в полученном биоптате. Синдромами с «рваными красными волокнами» являются следующие: MELAS, MERRF, KSS, PEO (прогрессирующая наружная офтальмоплегия), а также синдром Пирсона. Синдромы без RRF: болезнь Leigh, NARP, LHON (наследственная оптическая нейропатия Лебера) [2].
Генетические методы исследований сводятся к определению наиболее частых мутаций и секвенированию митохондриальной ДНК.
Лечение митохондриальной патологии
Терапия митохондриальных болезней, к сожалению, не разработана. С позиций доказательной медицины считается, что эффективное лечение для этой представительной группы болезней отсутствует. Тем не менее, в различных странах мира используются фармакологические средства и биологически активные вещества, нацеленные на нормализацию метаболизма и обеспечение адекватной энергетики митохондрий.
При синдроме MELAS лечение должно быть направлено на лечение судорог, эндокринных расстройств, устранение последствий инсульта.
P. Каufmann и соавт. (2006) указывают, что поскольку уровень лактата часто коррелирует с тяжестью неврологических проявлений, целесообразно применять дихлорацетат для снижения уровня лактата [11]. В нашей стране с аналогичной целью используется диметилоксобутилфосфонилдиметилат (Димефосфон) [12].
В исследованиях японских авторов Y. Koga и соавт. (2002, 2005, 2006, 2007) с хорошим эффектом использовалось внутривенное введение L-аргинина (предшественника NO) — для стимуляции вазодилатации в остром периоде инсульта, а также пероральное его применение для снижения тяжести последующих эпизодов [13–16].
Среди средств, используемых в терапии митохондриальной патологии, фигурируют следующие: витамин В1 (тиамин) — 400 мг/сут, витамин В2 (рибофлавин) — 100 мг/сут, витамин С (аскорбиновая кислота) — до 1 г/сут, витамин Е (токоферол) — 400 МЕ/сут, никотинамид (ниацин) — до 500 мг/сут, коэнзим Q10 — от 90 до 200 мг/сут, L-карнитин — от 10 мг до 1–2 г/сут, янтарная кислота — от 25 мг до 1,5 г/cут, Димефосфон 15% — 1,0 мл на 5 кг массы тела. Применяются также цитохром С (внутривенно), Реамберин (внутривенно) и Цитофлавин (внутривенно и перорально) [17, 18].
В качестве других средств фармакотерапии выступают кортикостероиды, минералокортикоиды (при развитии надпочечниковой недостаточности), антиконвульсанты — при судорогах/эпилепсии (исключая вальпроевую кислоту и ее производные, ограничивая применение барбитуратов). В наших наблюдениях наиболее эффективной противосудорожной терапией являлось использование препаратов леветирацетам (Кеппра), топирамат (Топамакс) или их сочетаний.
Нейродиетология при митохондриальной патологии
Основным принципом диеты при митохондриальной патологии является ограничение нутриентов, оказывающих негативное влияние на механизмы обмена — до формирования метаболического блока (рацион питания одновременно обогащается другими компонентами на обычном или повышенном уровне). Такая терапевтическая стратегия получила название «обхождения блока» (going around the block). Важным исключением в этом плане является группа митохондриальных нарушений, ассоциированных с метаболизмом пирувата (недостаточность пируватдегидрогеназного комплекса с сопутствующими нарушениями со стороны углеводов/гликогена/аминокислот). При этом рекомендуются кетогенная диета и другие виды высокожировых диет [19].
Широко применяются вещества, являющиеся пищевыми кофакторами (коэнзим Q10, L-карнитин, ацетил-L-карнитин, витамин В2, аскорбиновая кислота, витамин Е, витамин В1, никотинамид, витамин В6, витамин В12, биотин, фолиевая кислота, витамин К, α-липоевая кислота, янтарная кислота, Se) [19]. Рекомендуется избегание индивидуальных алиментарных факторов, индуцирующих обострение митохондриальной болезни (голодание, потребление жиров, белков, сахарозы, крахмала, алкоголя, кофеина, мононатрия глутамата; количественные нарушения приема пищи и неадекватное потребление пищевой энергии). При необходимости осуществляется клиническое питание (энтеральное, парентеральное, гастростомия) [19].
Чрезвычайно важными являются своевременная диагностика митохондриальных болезней, поиск клинических и параклинических критериев этих заболеваний на этапе предварительном, догенетическом. Это необходимо для подбора адекватной метаболической терапии и предотвращения ухудшения состояния или инвалидизации больных с этими редкими заболеваниями.
C. S. Chi (2015) подчеркивает, что подтверждение или исключение митохондриальной патологии остается принципиальным в педиатрической практике, особенно когда клинические признаки болезни не являются специфичными, вследствие чего необходим катамнестический подход к оценке симптомов и биохимических показателей [20].
Литература
* ГОУ ВПО РНИМУ им. Н. И. Пирогова МЗ РФ, Москва
** ГОУ ВПО ПМГМУ им. И. М. Сеченова МЗ РФ, Москва